Clear Sky Science · zh
用于药物-靶点结合亲和力预测的双分支图神经网络架构
更快通往更好药物的路径
开发一种新药通常需要十多年和数十亿美元投入,只有极少数候选药物最终能够进入临床应用。此项研究探讨了一种新型人工智能——将分子视为连接原子的网络——如何加速药物筛选,甚至为现有药物发现新的用途。通过预测药物与体内蛋白靶点的结合强度,该方法旨在把成千上万的可能性缩减为可操作的高质量候选列表。
为何预测“粘性”很重要
大多数现代药物通过与特定蛋白结合并改变其功能来发挥作用。结合强度(binding affinity)与药物是否有效以及所需剂量密切相关。传统上,测定结合亲和力需要耗时的实验室工作和大量化合物库。作者们着重用计算方法替代这部分反复试验:如果计算模型能够可靠地预测哪些药物—蛋白对可能紧密结合,研究者就能在实验室中测试更少的化合物,从而节省时间和资金,同时为新的治疗思路打开大门。
将分子视为互联的地图
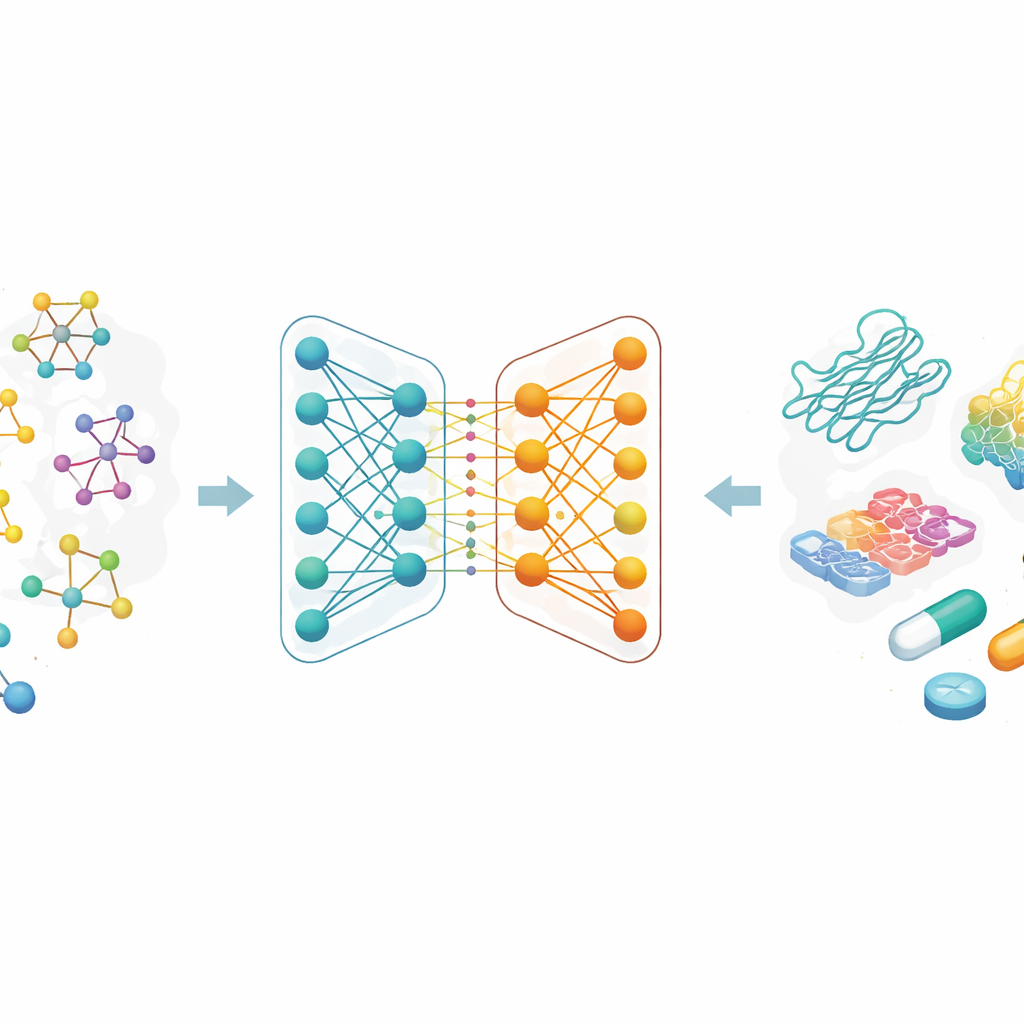
研究人员没有把分子描述为字母串或冗长的手工特征列表,而是将每种药物视为一个图:原子是节点,化学键是连接线。被称为图神经网络的一类人工智能方法特别善于从这些地图中学习,因为它们沿着连接反复传递信息,既能捕捉局部细节又能把握整体结构。在这项工作中,每个药物从化学中常用的类似文本的编码开始,然后转换为分子图。研究团队还为蛋白靶点设计了丰富的编码,旨在捕捉氨基酸序列中的模式及具有化学意义的子结构。
协同工作的两条 AI 分支
论文的核心是一种新的用于药物表征的双分支图神经网络。一个分支使用图卷积网络(graph convolutional network),以受控的方式融合相邻原子的信号;另一个分支使用称为 GraphSAGE 的方法,从数据中学习如何概括每个原子的邻域信息。随后,一个“跳跃式知识”(jumping knowledge)模块灵活地结合来自不同深度的信息,使最终的药物表征既反映浅层的局部模式,又包含更深层的全局特征。这一双分支编码与强大的蛋白编码器配对,所得的药物—蛋白特征被输入到预测模块,输出估计的结合强度。
模型的表现如何
为检验其设计,作者将该模型与45种不同的现有药物和蛋白编码器组合在两组广泛使用的基准数据集上进行了比较:Davis 和 KIBA 数据集,这两组数据记录了多种小分子与蛋白激酶(一类重要的药物靶点)之间的相互作用。在多项常用的准确性和排序质量指标上,新的双分支模型始终位于前列或接近领先位置。尤其是在 Davis 数据集上,它比其他基于图的方法提供了更低的预测误差和更优的药物—靶点排序,这表明其对分子结构的更丰富表达捕捉到了更为细微的差异。在规模更大且噪声更高的 KIBA 数据集中,改进幅度较小但仍然稳健,显示了良好的泛化能力。
从数据到真实疾病案例
为展示实际价值,团队将模型用于新冠病毒(COVID-19)药物重定位的案例研究,聚焦许多治疗旨在抑制的主要病毒酶。人工智能按预测的结合强度对现有抗病毒药物及相关药物进行排序。若干知名候选药物(包括瑞德西韦和其他蛋白酶抑制剂)出现在排名前列,与其他研究小组报告的实验结果一致。当将模型的排序与传统计算对接(docking)得分比较时,两者高度一致,且该 AI 在把可能的命中推到前列方面表现出特别的优势。这表明此类模型可作为强有力的筛选工具,在快速变化的公共卫生危机中优先筛选待实验验证的化合物。
对未来药物发现的意义
总体而言,该研究表明精心设计的基于图的人工智能能够为药物分子提供更具信息量的指纹并更可靠地预测其与蛋白靶点的相互作用。对非专业读者而言,核心信息是:更智能的化学数字表示可以使虚拟筛选更准确、更可行,尤其在时间和资源有限的情况下。尽管该方法仍面临挑战——包括计算成本和对设计选择的敏感性——它代表了朝着能帮助科学家更高效、更安全地重定位现有药物和设计新药的人工智能系统迈出的重要一步。
引用: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
关键词: 图神经网络, 药物发现, 结合亲和力预测, 药物重定位, 计算药理学