Clear Sky Science · en
A dual-branch graph neural network architecture for drug-target binding affinity prediction
Faster paths to better medicines
Developing a new drug usually takes more than a decade and billions of dollars, with only a small fraction of candidates ever reaching patients. This study explores how a new kind of artificial intelligence, built to understand molecules as networks of connected atoms, can speed up the search for drugs and even find fresh uses for existing medicines. By predicting how strongly a drug will stick to a protein target in the body, the approach aims to narrow thousands of possibilities down to a manageable, high-quality shortlist.
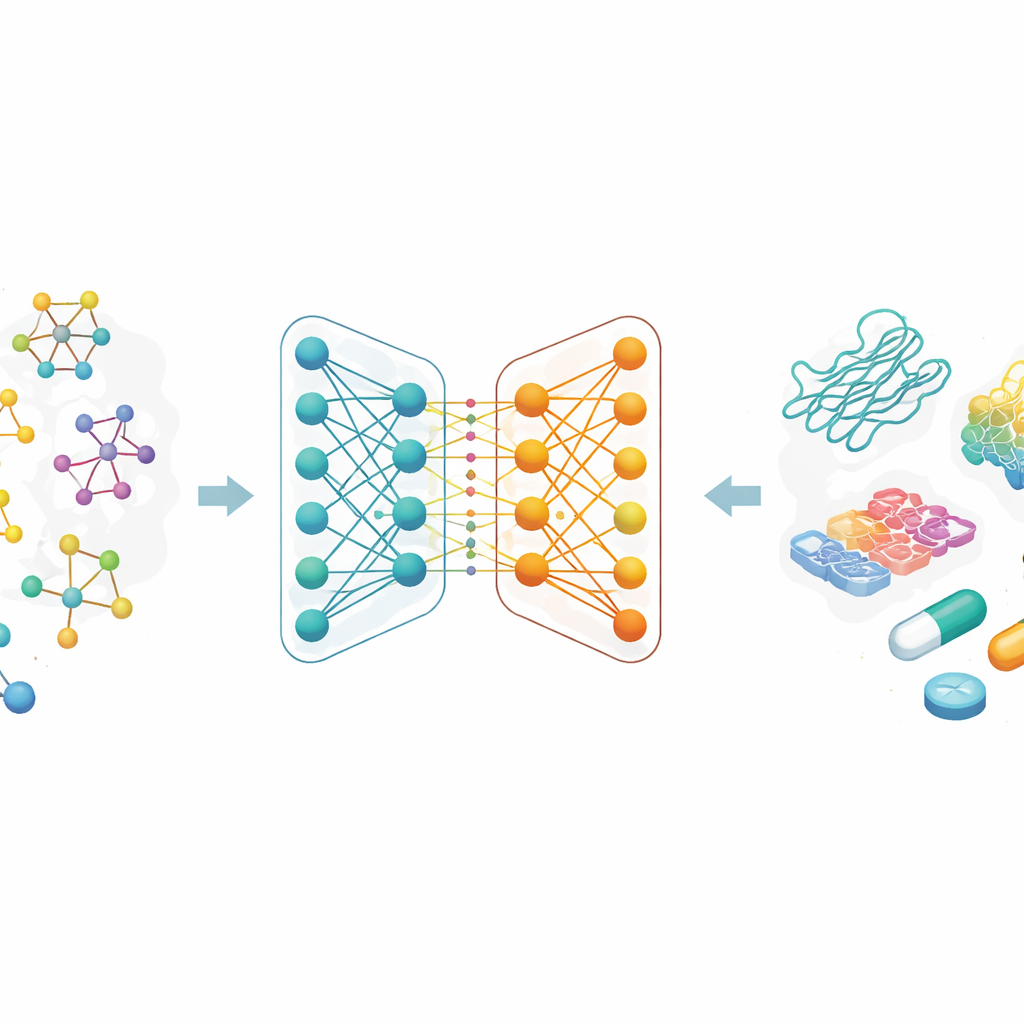
Why predicting stickiness matters
Most modern drugs work by latching onto specific proteins and changing how they behave. The strength of that grip, called binding affinity, is closely tied to whether a drug will work and at what dose. Traditionally, measuring binding affinity requires laborious lab experiments and large libraries of chemical compounds. The authors focus on replacing a portion of this trial-and-error process with computation: if a computer model can reliably predict which drug–protein pairs are likely to bind tightly, researchers can test far fewer compounds in the lab, saving both time and money while opening the door to new treatment ideas.
Seeing molecules as interconnected maps
Instead of describing molecules as strings of letters or long lists of handcrafted features, the researchers treat each drug as a graph: atoms are points, and chemical bonds are lines between them. A family of AI methods called graph neural networks is particularly good at learning from these maps, because it repeatedly passes information along the connections, capturing both local details and global structure. In this work, each drug starts from a common text-like code used in chemistry, which is then converted into a molecular graph. The team also uses rich encodings for protein targets, designed to capture patterns in amino acid sequences and chemically meaningful substructures.
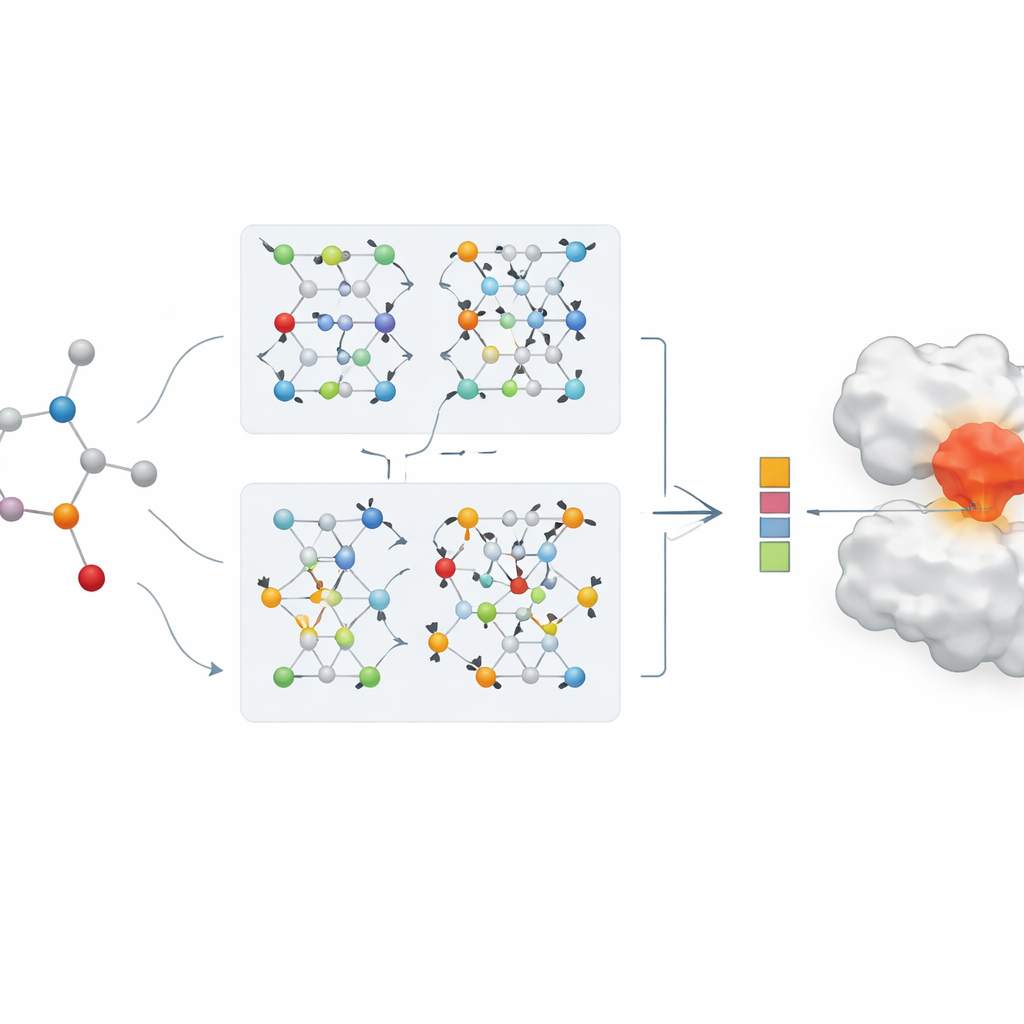
Two AI branches working together
The heart of the paper is a new dual-branch graph neural network for drug representation. One branch uses a graph convolutional network, which blends information from neighboring atoms in a controlled way. The other uses a method called GraphSAGE, which learns how to summarize each atom’s neighborhood from data. A “jumping knowledge” module then flexibly combines information from different depths of the network, so the final drug representation reflects both shallow, local patterns and deeper, more global ones. This dual-branch encoding is paired with a strong protein encoder, and the combined drug–protein features are fed into a prediction module that outputs an estimated binding strength.
How well the model performs
To test their design, the authors compare it against 45 different combinations of existing drug and protein encoders on two widely used benchmark collections: the Davis and KIBA datasets, which catalog how a variety of small molecules interact with protein kinases, an important class of drug targets. Across several standard measures of accuracy and ranking quality, the new dual-branch model consistently sits at or near the top. On the Davis dataset in particular, it delivers lower prediction error and better ordering of drug–target pairs than competing graph-based methods, suggesting that its richer view of molecular structure captures subtle differences that simpler models miss. On the larger and noisier KIBA dataset, improvements are smaller but still robust, indicating good generalization.
From numbers to real disease cases
To demonstrate real-world value, the team applies their model to a drug-repurposing case study for COVID-19, focusing on the main viral enzyme that many treatments aim to disable. The AI ranks existing antiviral and related drugs by predicted binding strength to this enzyme. Several well-known candidates, including remdesivir and other protease inhibitors, appear near the top of the list, aligning with experimental findings reported by other groups. When the model’s rankings are compared with traditional computer docking scores, there is strong agreement, and the AI shows particular strength in pushing likely hits toward the top. This suggests that such models could serve as powerful filters to prioritize compounds for experimental testing during fast-moving health crises.
What this means for future drug discovery
Overall, the study shows that carefully designed graph-based AI can provide more informative fingerprints of drug molecules and more reliable predictions of how they interact with protein targets. For non-specialists, the key message is that smarter digital representations of chemistry can make virtual screening more accurate and practical, especially when time and resources are limited. While the approach still faces challenges—including computational cost and sensitivity to design choices—it represents a meaningful step toward AI systems that help scientists repurpose existing drugs and design new ones more efficiently and safely.
Citation: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Keywords: graph neural networks, drug discovery, binding affinity prediction, drug repurposing, computational pharmacology