Clear Sky Science · ru
Архитектура графовой нейронной сети с двумя ветвями для предсказания аффинности связывания лекарство–мишень
Более быстрые пути к лучшим лекарствам
Разработка нового препарата обычно занимает более десяти лет и требует миллиардов долларов, при этом лишь небольшая часть кандидатов когда‑либо доходит до пациентов. В этом исследовании рассматривается, как новый тип искусственного интеллекта, созданный для понимания молекул как сетей связанных атомов, может ускорить поиск лекарств и даже найти новые применения для существующих препаратов. Предсказывая, насколько сильно лекарство будет «прилипать» к белковой мишени в организме, подход нацелен на то, чтобы сузить тысячи вариантов до управляемого списка высокого качества.
Почему важно предсказывать «липкость»
Большинство современных лекарств действуют, присоединяясь к определённым белкам и изменяя их поведение. Сила этой связи, называемая аффинностью связывания, тесно связана с тем, будет ли препарат эффективен и в какой дозировке. Традиционно измерение аффинности требует трудоёмких лабораторных экспериментов и больших библиотек химических соединений. Авторы сосредотачиваются на замене части этого метода проб и ошибок вычислениями: если модель сможет надёжно предсказывать, какие пары «лекарство–белок» скорее всего будут сильно связываться, исследователи смогут протестировать в лаборатории гораздо меньше соединений, экономя время и деньги и открывая путь к новым идеям для лечения.
Видеть молекулы как взаимосвязанные карты
Вместо описания молекул строками символов или длинными списками вручную подобранных признаков, исследователи рассматривают каждое лекарство как граф: атомы — это узлы, а химические связи — рёбра между ними. Семейство методов ИИ под названием графовые нейронные сети особенно хорошо обучается на таких картах, поскольку многократно передаёт информацию по связям, захватывая как локальные детали, так и глобальную структуру. В работе каждое соединение сначала представлено в виде привычного текстового химического кода, который затем преобразуют в молекулярный граф. Команда также использует богатые кодировки для белковых мишеней, предназначенные для улавливания шаблонов в последовательностях аминокислот и химически значимых субструктур.
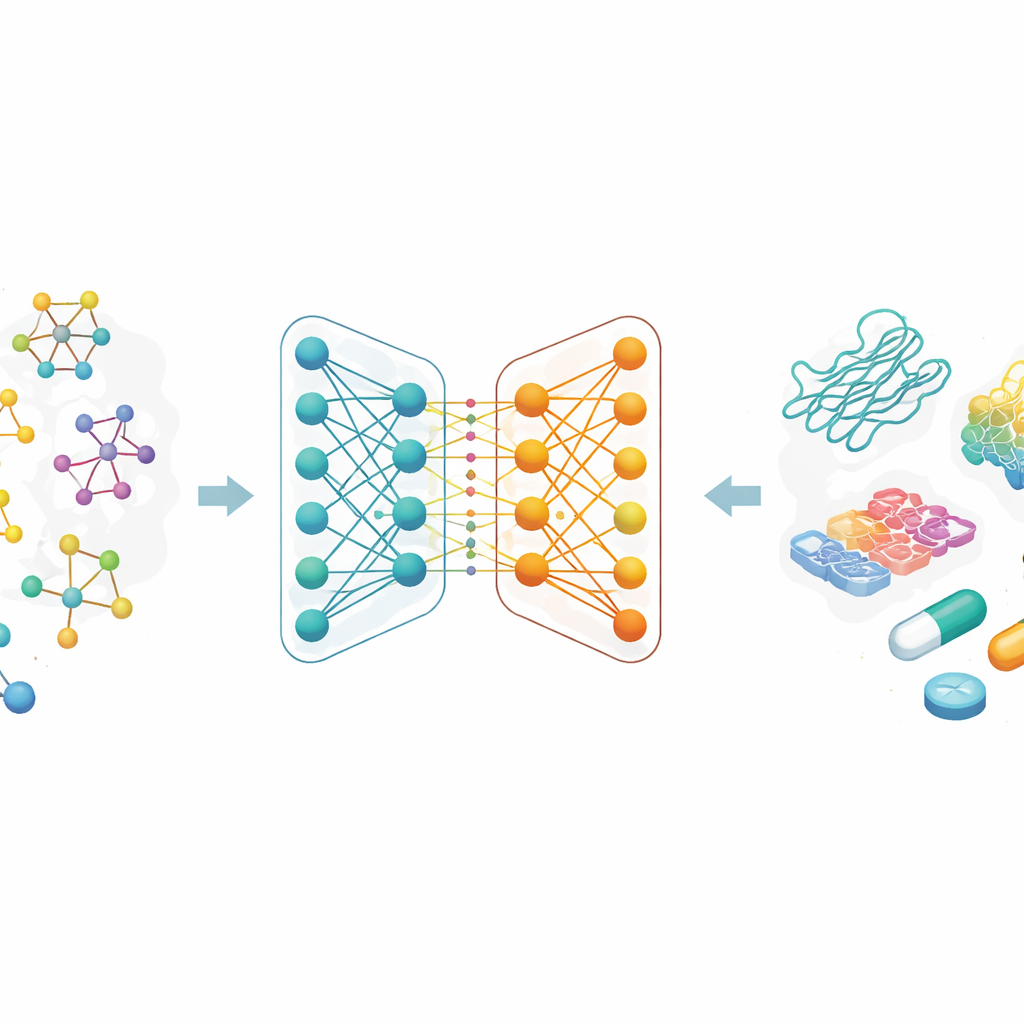
Две ветви ИИ, работающие вместе
В основе статьи лежит новая графовая нейронная сеть с двумя ветвями для представления лекарств. Одна ветвь использует графовую сверточную сеть, которая контролируемо объединяет информацию от соседних атомов. Другая применяет метод GraphSAGE, который учится суммировать окрестность каждого атома на основе данных. Модуль «прыгающего знания» затем гибко комбинирует информацию с разных глубин сети, так что окончательное представление лекарства отражает и поверхностные локальные шаблоны, и более глубокие глобальные особенности. Это двуветвевое кодирование сочетается с мощным энкодером белка, а объединённые признаки «лекарство–белок» подаются в модуль предсказания, выдающий оценку силы связывания.
Насколько хорошо работает модель
Чтобы оценить свою архитектуру, авторы сравнивают её с 45 различными сочетаниями существующих энкодеров для лекарств и белков на двух широко используемых бенчмарках: наборах данных Davis и KIBA, которые каталогизируют взаимодействия множества малых молекул с киназами белков — важным классом мишеней. По нескольким стандартным метрикам точности и качества ранжирования новая двуветвеевая модель стабильно занимает верхние позиции или близки к ним. В частности, на наборе Davis она демонстрирует меньшую ошибку предсказания и лучшее ранжирование пар «лекарство–мишень» по сравнению с конкурирующими графовыми методами, что говорит о том, что её более богатое представление молекулярной структуры улавливает тонкие различия, которые пропускают более простые модели. На более крупном и более шумном наборе KIBA улучшения меньше, но всё ещё стойки, указывая на хорошую обобщающую способность.
От чисел к реальным случаям заболеваний
Чтобы продемонстрировать практическую ценность, команда применяет свою модель в кейсе переназначения лекарств для COVID-19, сосредоточившись на главном вирусном ферменте, который многие препараты стремятся отключить. ИИ ранжирует существующие противовирусные и родственные препараты по предсказанной силе связывания с этим ферментом. Несколько хорошо известных кандидатов, включая ремдесивир и другие ингибиторы протеаз, оказываются в верхней части списка, что согласуется с экспериментальными результатами, опубликованными другими группами. При сравнении ранжирования модели с традиционными оценками докинга наблюдается сильное совпадение, причём ИИ особенно хорошо поднимает вероятные «попадания» в начало списка. Это предполагает, что такие модели могут служить мощными фильтрами для приоритизации соединений для экспериментальной проверки в быстро меняющихся кризисах здравоохранения.
Что это значит для будущего поиска лекарств
В целом исследование показывает, что тщательно продуманные графовые методы ИИ могут давать более информативные «отпечатки» молекул лекарств и более надёжные предсказания их взаимодействия с белковыми мишенями. Для неспециалистов ключевое сообщение таково: более умные цифровые представления химии могут сделать виртуальный скрининг точнее и практичнее, особенно когда время и ресурсы ограничены. Хотя подход всё ещё сталкивается с трудностями — включая вычислительные затраты и чувствительность к выбору архитектуры — он представляет собой значимый шаг к системам ИИ, которые помогают учёным переназначать существующие препараты и разрабатывать новые более эффективно и безопасно.
Цитирование: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Ключевые слова: графовые нейронные сети, поиск лекарств, предсказание аффинности связывания, переназначение лекарств, вычислительная фармакология