Clear Sky Science · nl
Een duale tak-architectuur van graph neural networks voor voorspelling van bindingaffiniteit tussen geneesmiddel en doelwit
Snellere routes naar betere medicijnen
Het ontwikkelen van een nieuw geneesmiddel duurt gewoonlijk meer dan tien jaar en kost miljarden dollars, waarbij slechts een klein deel van de kandidaten uiteindelijk bij patiënten terechtkomt. Deze studie onderzoekt hoe een nieuw type kunstmatige intelligentie, gebouwd om moleculen te begrijpen als netwerken van verbonden atomen, het zoeken naar geneesmiddelen kan versnellen en zelfs nieuwe toepassingen voor bestaande medicijnen kan vinden. Door te voorspellen hoe sterk een geneesmiddel aan een eiwitdoelwit in het lichaam zal hechten, probeert de methode duizenden mogelijkheden terug te brengen tot een beheersbare, kwalitatief hoogwaardige short‑list.
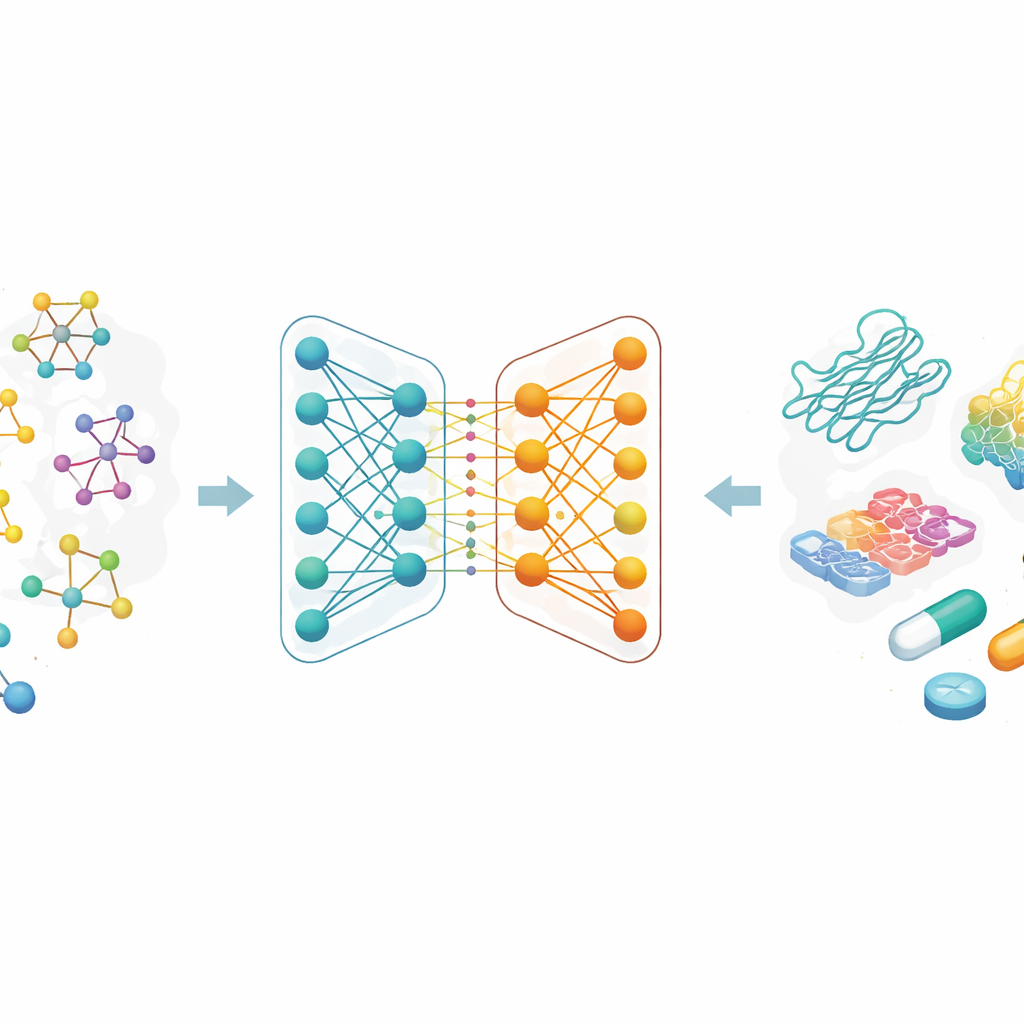
Waarom het voorspellen van hechten belangrijk is
De meeste moderne geneesmiddelen werken door zich vast te hechten aan specifieke eiwitten en hun gedrag te veranderen. De sterkte van die binding, bindingaffiniteit genoemd, hangt nauw samen met of een geneesmiddel zal werken en bij welke dosis. Traditioneel vereist het meten van bindingaffiniteit arbeidsintensieve laboratoriumexperimenten en grote bibliotheken met chemische verbindingen. De auteurs richten zich op het vervangen van een deel van dit trial‑and‑error‑proces door berekeningen: als een computermodel betrouwbaar kan voorspellen welke geneesmiddel‑eiwitparen waarschijnlijk sterk binden, kunnen onderzoekers veel minder verbindingen in het laboratorium testen, wat tijd en geld bespaart en nieuwe behandelideeën mogelijk maakt.
Moleculen zien als onderling verbonden kaarten
In plaats van moleculen te beschrijven als letterreeksen of lange lijsten handgemaakte kenmerken, behandelen de onderzoekers elk geneesmiddel als een graaf: atomen zijn punten en chemische bindingen lijnen daartussen. Een familie van AI-methoden, graph neural networks, is bijzonder goed in het leren van zulke kaarten, omdat ze herhaaldelijk informatie langs de verbindingen sturen en zo zowel lokale details als globale structuur vastleggen. In dit werk begint elk geneesmiddel met een veelgebruikt tekstachtig chemisch codeformaat, dat vervolgens wordt omgezet in een moleculaire graaf. Het team gebruikt ook rijke encoderingen voor eiwitdoelwitten, ontworpen om patronen in aminozuursequensen en chemisch betekenisvolle substructuren te vatten.
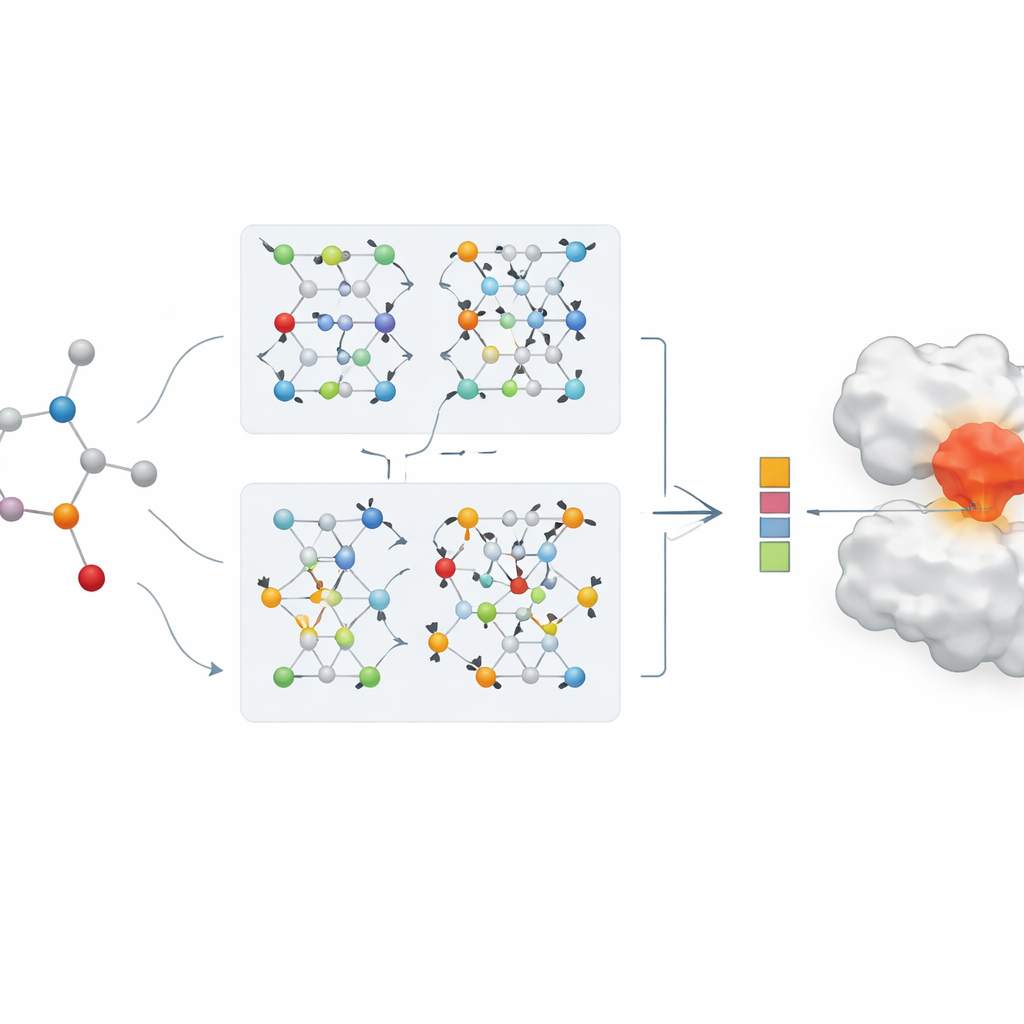
Twee AI-takken die samenwerken
Het hart van het artikel is een nieuwe duale tak van graph neural networks voor geneesmiddelrepresentatie. De ene tak gebruikt een graph convolutional network, dat informatie van naburige atomen op een gecontroleerde manier mengt. De andere gebruikt een methode genaamd GraphSAGE, die leert hoe de buurt van elk atoom uit gegevens samen te vatten. Een "jumping knowledge"-module combineert vervolgens flexibel informatie van verschillende dieptes van het netwerk, zodat de uiteindelijke geneesmiddelrepresentatie zowel ondiepe, lokale patronen als diepere, meer globale structuren weerspiegelt. Deze duale codering wordt gekoppeld aan een krachtig eiwit‑encoder, en de gecombineerde geneesmiddel‑eiwitkenmerken worden gevoed aan een predictiemodule die een geschatte bindingssterkte afgeeft.
Hoe goed het model presteert
Om hun ontwerp te testen vergelijken de auteurs het met 45 verschillende combinaties van bestaande geneesmiddel‑ en eiwitencoders op twee veelgebruikte benchmarkverzamelingen: de Davis en KIBA datasets, die vastleggen hoe een verscheidenheid aan kleine moleculen interageert met proteïnekinasen, een belangrijke klasse van geneesmiddeldoelen. Over meerdere standaardmaatstaven voor nauwkeurigheid en rangschikkingskwaliteit staat het nieuwe duale model consequent bovenaan of in de buurt daarvan. Vooral op de Davis‑dataset levert het lagere voorspellingsfout en betere ordening van geneesmiddel‑doelparen dan concurrerende grafgebaseerde methoden, wat suggereert dat zijn rijkere kijk op moleculaire structuur subtiele verschillen vastlegt die eenvoudigere modellen missen. Op de grotere en luidruchtigere KIBA‑dataset zijn de verbeteringen kleiner maar nog steeds robuust, wat duidt op goede generalisatie.
Van cijfers naar echte ziektegevallen
Om de waarde in de echte wereld aan te tonen past het team hun model toe op een case study voor herbestemming van geneesmiddelen bij COVID‑19, met focus op het belangrijke virale enzym dat veel behandelingen tot doel hebben uit te schakelen. De AI rangschikt bestaande antivirale en verwante geneesmiddelen naar voorspelde bindingssterkte aan dit enzym. Verschillende bekende kandidaten, waaronder remdesivir en andere proteaseremmers, verschijnen hoog in de lijst en komen overeen met experimentele bevindingen van andere groepen. Wanneer de ranking van het model wordt vergeleken met traditionele computer‑docking scores is er sterke overeenstemming, en toont de AI bijzondere kracht in het naar boven duwen van waarschijnlijke hits. Dit suggereert dat zulke modellen als krachtige filters kunnen dienen om verbindingen te prioriteren voor experimentele testen tijdens snel bewegende gezondheidscrisissen.
Wat dit betekent voor toekomstig geneesmiddelenonderzoek
Al met al laat de studie zien dat zorgvuldig ontworpen graafgebaseerde AI meer informatieve vingerafdrukken van geneesmiddelmoleculen kan bieden en betrouwbaardere voorspellingen van hoe ze met eiwitdoelen interageren. Voor niet‑specialisten is de kernboodschap dat slimmer digitale representaties van chemie virtueel screenen nauwkeuriger en praktischer kunnen maken, vooral wanneer tijd en middelen beperkt zijn. Hoewel de aanpak nog uitdagingen kent — waaronder rekenkosten en gevoeligheid voor ontwerpkeuzes — vormt ze een betekenisvolle stap richting AI‑systemen die wetenschappers helpen bestaande geneesmiddelen herbestemmen en nieuwe efficiënter en veiliger te ontwerpen.
Bronvermelding: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Trefwoorden: graph neural networks, geneesmiddelenonderzoek, voorspelling van bindingaffiniteit, herbestemming van geneesmiddelen, computationele farmacologie