Clear Sky Science · fr
Une architecture de réseau de neurones graphe à double branche pour la prédiction de l'affinité de liaison médicament-cible
Des voies plus rapides vers de meilleurs médicaments
Développer un nouveau médicament prend généralement plus d'une décennie et coûte des milliards de dollars, et seule une faible fraction des candidats atteint les patients. Cette étude explore comment un nouveau type d'intelligence artificielle, conçu pour comprendre les molécules comme des réseaux d'atomes connectés, peut accélérer la recherche de médicaments et même identifier de nouvelles utilisations pour des médicaments existants. En prédisant dans quelle mesure un médicament adhère à une protéine cible dans l'organisme, l'approche vise à réduire des milliers de possibilités à une liste restreinte gérable et de haute qualité.
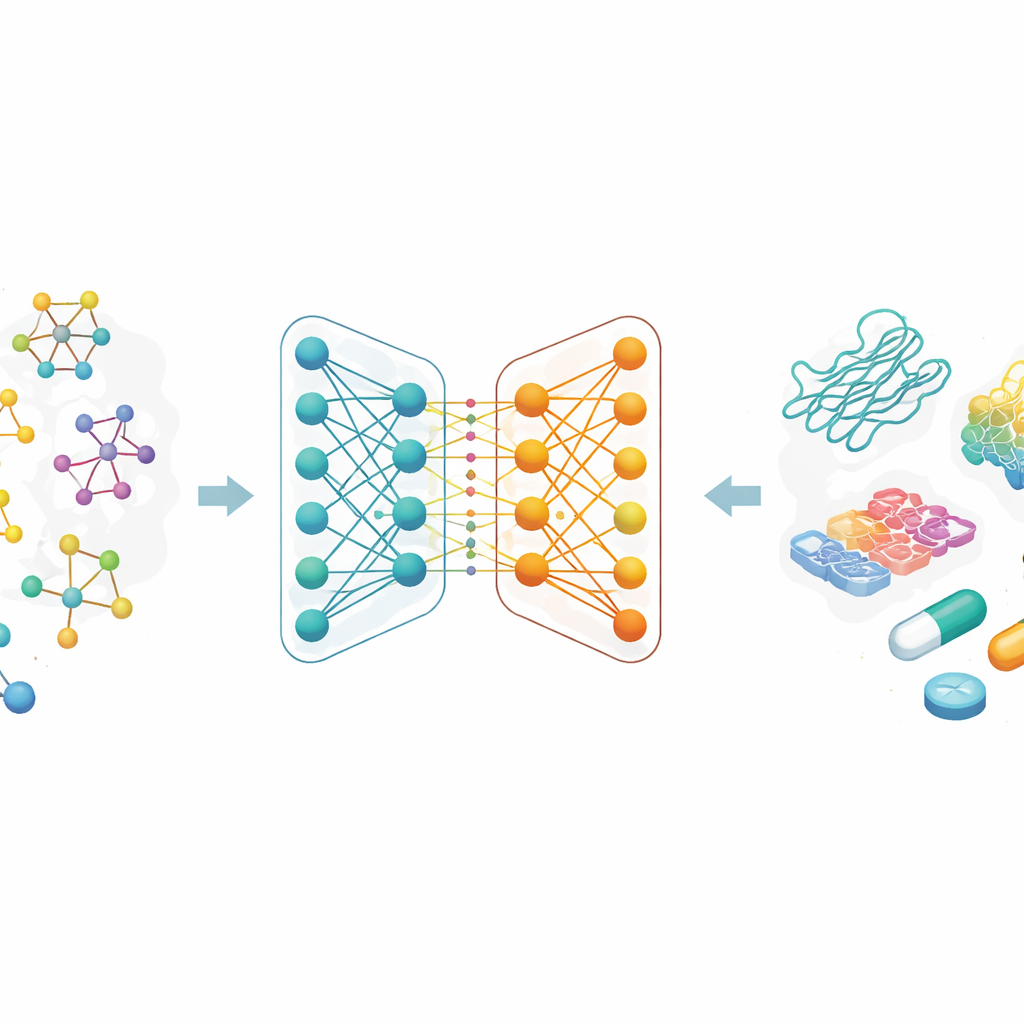
Pourquoi la prédiction de l'adhérence est importante
La plupart des médicaments modernes agissent en se liant à des protéines spécifiques et en modifiant leur comportement. La force de cette liaison, appelée affinité de liaison, est étroitement liée à l'efficacité d'un médicament et à la dose nécessaire. Traditionnellement, mesurer l'affinité de liaison exige des expériences laborieuses en laboratoire et de grandes bibliothèques de composés chimiques. Les auteurs cherchent à remplacer une partie de ce processus d'essais-erreurs par des calculs : si un modèle informatique peut prédire de manière fiable quelles paires médicament–protéine sont susceptibles de se lier fortement, les chercheurs peuvent tester beaucoup moins de composés en laboratoire, économisant temps et argent tout en ouvrant la voie à de nouvelles idées thérapeutiques.
Voir les molécules comme des cartes interconnectées
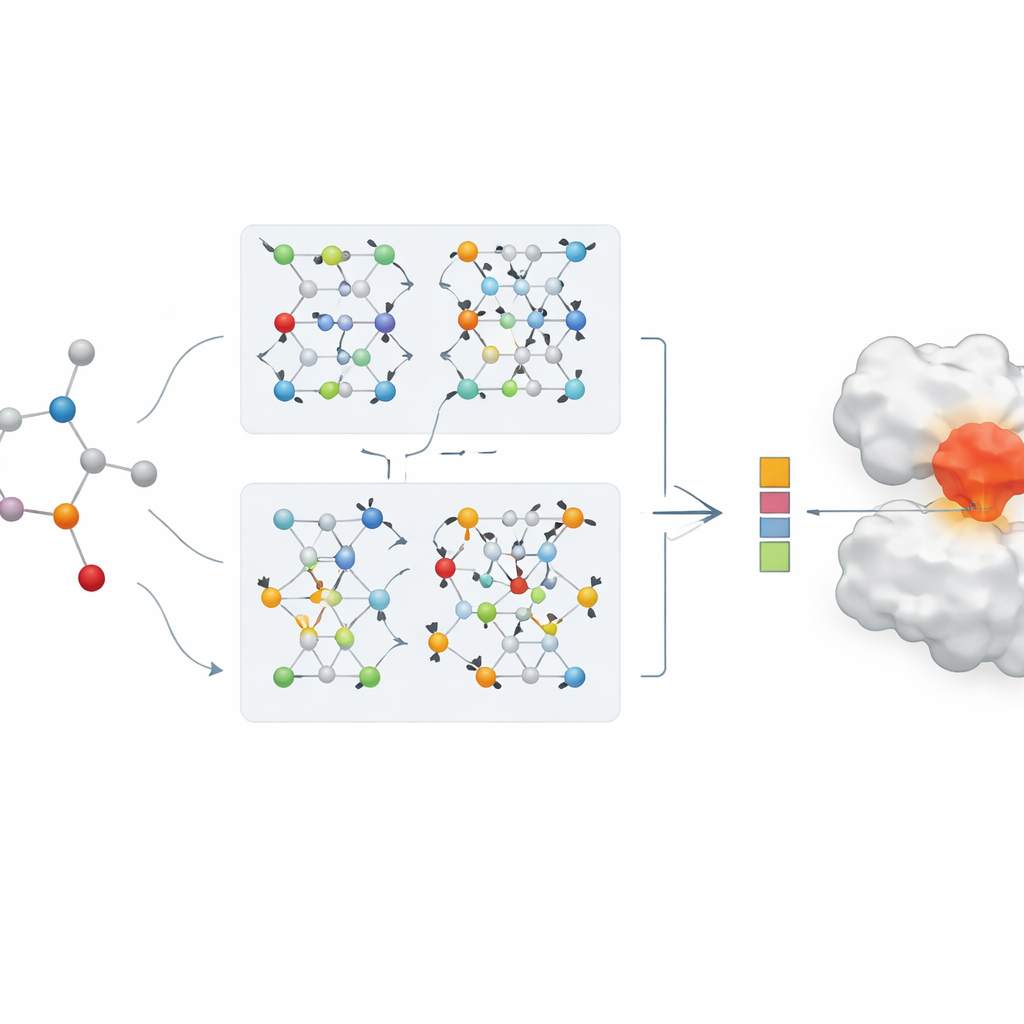
Plutôt que de décrire les molécules comme des chaînes de lettres ou de longues listes de caractéristiques fabriquées à la main, les chercheurs considèrent chaque médicament comme un graphe : les atomes sont des nœuds et les liaisons chimiques des arêtes entre eux. Une famille de méthodes d'IA appelée réseaux de neurones graphe est particulièrement adaptée pour apprendre à partir de ces cartes, car elle fait circuler l'information le long des connexions, capturant à la fois les détails locaux et la structure globale. Dans ce travail, chaque médicament part d'un code textuel courant en chimie, qui est ensuite converti en un graphe moléculaire. L'équipe utilise également des encodages riches pour les protéines cibles, conçus pour capturer des motifs dans les séquences d'acides aminés et des sous-structures chimiquement significatives.
Deux branches d'IA qui travaillent ensemble
Le cœur de l'article est un nouveau réseau de neurones graphe à double branche pour la représentation des médicaments. Une branche utilise un réseau de convolution sur graphe, qui mélange l'information provenant des atomes voisins de manière contrôlée. L'autre utilise une méthode appelée GraphSAGE, qui apprend à résumer le voisinage de chaque atome à partir des données. Un module de « jumping knowledge » combine ensuite de façon flexible l'information provenant de différentes profondeurs du réseau, de sorte que la représentation finale du médicament reflète à la fois des motifs superficiels et locaux et des motifs plus profonds et globaux. Cet encodage à double branche est associé à un encodeur de protéines robuste, et les caractéristiques combinées médicament–protéine sont transmises à un module de prédiction qui fournit une estimation de la force de liaison.
Performance du modèle
Pour évaluer leur conception, les auteurs la comparent à 45 combinaisons différentes d'encodeurs médicament et protéine existants sur deux collections de référence largement utilisées : les jeux de données Davis et KIBA, qui répertorient comment une variété de petites molécules interagissent avec les kinases protéiques, une classe importante de cibles médicamenteuses. Sur plusieurs mesures standard d'exactitude et de qualité de classement, le nouveau modèle à double branche se situe systématiquement au sommet ou proche du sommet. Sur le jeu de données Davis en particulier, il délivre une erreur de prédiction plus faible et un meilleur ordonnancement des paires médicament–cible que les méthodes concurrentes basées sur les graphes, suggérant que sa vision plus riche de la structure moléculaire capte des différences subtiles que des modèles plus simples manquent. Sur le jeu de données plus grand et plus bruité KIBA, les améliorations sont plus modestes mais restent solides, indiquant une bonne capacité de généralisation.
Des chiffres aux cas de maladie réels
Pour démontrer la valeur en conditions réelles, l'équipe applique son modèle à une étude de cas de réaffectation de médicaments pour la COVID-19, en se concentrant sur l'enzyme virale principale que de nombreux traitements cherchent à désactiver. L'IA classe les antiviraux existants et les médicaments apparentés selon la force de liaison prédite à cette enzyme. Plusieurs candidats bien connus, y compris le remdésivir et d'autres inhibiteurs de protéase, apparaissent en haut de la liste, en accord avec des résultats expérimentaux rapportés par d'autres équipes. Lorsque les classements du modèle sont comparés aux scores traditionnels de docking informatique, il existe une forte concordance, et l'IA montre une capacité particulière à faire remonter les candidats probables en tête. Cela suggère que de tels modèles pourraient servir de filtres puissants pour prioriser les composés à tester expérimentalement lors de crises sanitaires rapides.
Ce que cela signifie pour la découverte de médicaments à venir
Dans l'ensemble, l'étude montre que des IA basées sur les graphes et soigneusement conçues peuvent fournir des empreintes numériques plus informatives des molécules médicamenteuses et des prédictions plus fiables de leur interaction avec des protéines cibles. Pour les non-spécialistes, le message clé est que des représentations numériques plus intelligentes de la chimie peuvent rendre le criblage virtuel plus précis et plus pratique, surtout lorsque le temps et les ressources sont limités. Si l'approche doit encore surmonter des défis — y compris le coût computationnel et la sensibilité aux choix de conception — elle représente une avancée significative vers des systèmes d'IA qui aident les scientifiques à réaffecter des médicaments existants et à concevoir de nouveaux médicaments de manière plus efficace et plus sûre.
Citation: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Mots-clés: réseaux de neurones graphe, découverte de médicaments, prédiction de l'affinité de liaison, réaffectation de médicaments, pharmacologie computationnelle