Clear Sky Science · sv
En tvågrenad grafneural nätverksarkitektur för prediktion av läkemedels–målets bindningsaffinitet
Snabbare vägar till bättre läkemedel
Att utveckla ett nytt läkemedel tar vanligtvis mer än ett decennium och kostar miljarder dollar, och endast en liten andel kandidater når någonsin patienter. Denna studie undersöker hur en ny typ av artificiell intelligens, byggd för att förstå molekyler som nätverk av sammanlänkade atomer, kan påskynda sökandet efter läkemedel och till och med hitta nya användningsområden för befintliga mediciner. Genom att förutsäga hur starkt ett läkemedel kommer att binda till ett proteintarget i kroppen syftar metoden till att krympa tusentals möjligheter till en hanterbar, högkvalitativ shortlist.
Varför bindningsstyrka spelar roll
De flesta moderna läkemedel verkar genom att fästa vid specifika proteiner och ändra deras beteende. Styrkan i det greppet, kallad bindningsaffinitet, är nära kopplad till om ett läkemedel kommer att fungera och vid vilken dos. Traditionellt kräver mätning av bindningsaffinitet mödosamma laboratorieexperiment och stora bibliotek av kemiska föreningar. Författarna fokuserar på att ersätta en del av denna trial-and-error-process med beräkningar: om en datormodell pålitligt kan förutsäga vilka läkemedels–proteinpar som sannolikt binder starkt, kan forskare testa långt färre föreningar i labbet, spara både tid och pengar och öppna dörren för nya behandlingsidéer.
Att se molekyler som sammankopplade kartor
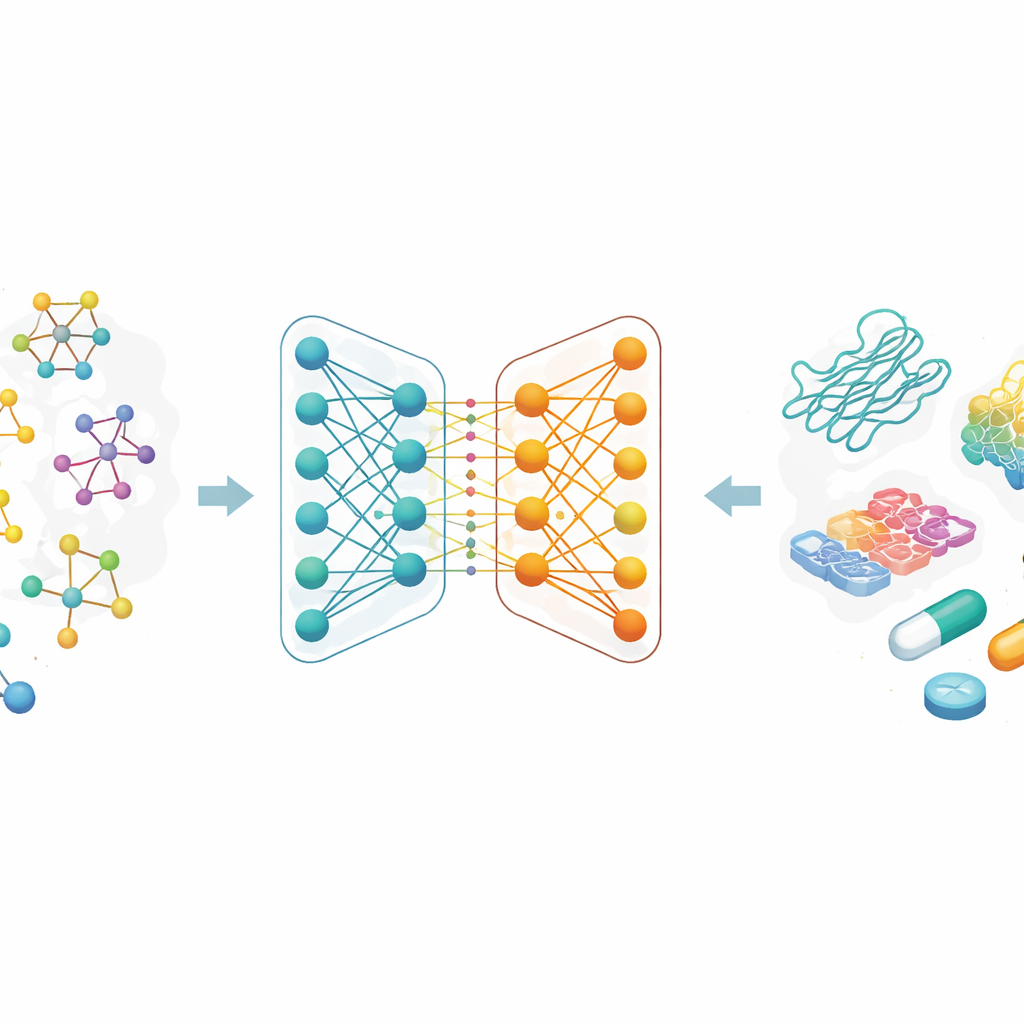
I stället för att beskriva molekyler som bokstavssträngar eller långa listor av handgjorda egenskaper behandlar forskarna varje läkemedel som en graf: atomer är punkter och kemiska bindningar är linjer mellan dem. En familj av AI-metoder kallad grafneuralnätverk är särskilt bra på att lära från dessa kartor, eftersom de upprepade gånger skickar information längs kopplingarna och fångar både lokala detaljer och global struktur. I detta arbete utgår varje läkemedel från en vanlig textlik kod som används i kemi, vilken sedan konverteras till en molekylär graf. Teamet använder också rika kodningar för proteintargets, utformade för att fånga mönster i aminosyrasekvenser och kemiskt meningsfulla substrukturer.
Två AI-grenar som samarbetar
Kärnan i artikeln är ett nytt tvågrenat grafneuralnätverk för representation av läkemedel. Den ena grenen använder ett grafkonvolutionsnätverk, som blandar information från närliggande atomer på ett kontrollerat sätt. Den andra använder en metod kallad GraphSAGE, som lär sig hur man summerar varje atoms grannskap från data. En "jumping knowledge"-modul kombinerar sedan flexibelt information från olika djup i nätverket, så att den slutliga läkemedelsrepresentationen speglar både ytliga, lokala mönster och djupare, mer globala sådana. Denna tvågrenade kodning paras med en kraftfull proteinkodare, och de kombinerade läkemedels–proteinfunktionerna matas in i en prediktionsmodul som ger en uppskattad bindningsstyrka.
Hur bra modellen presterar
För att testa sin design jämför författarna den med 45 olika kombinationer av befintliga läkemedels- och proteinkodare på två allmänt använda benchmark-samlingar: Davis- och KIBA-datamängderna, som katalogiserar hur en mängd små molekyler interagerar med proteinkinaser, en viktig klass av läkemedelsmål. Över flera standardmått för noggrannhet och rankningskvalitet ligger den nya tvågrenade modellen konsekvent i toppen eller nära toppen. Särskilt på Davis-datamängden levererar den lägre prediktionsfel och bättre ordning av läkemedels–målpar än konkurrerande grafbaserade metoder, vilket tyder på att dess rikare bild av molekylär struktur fångar subtila skillnader som enklare modeller missar. På den större och mer brusiga KIBA-datamängden är förbättringarna mindre men fortfarande robusta, vilket indikerar god generalisering.
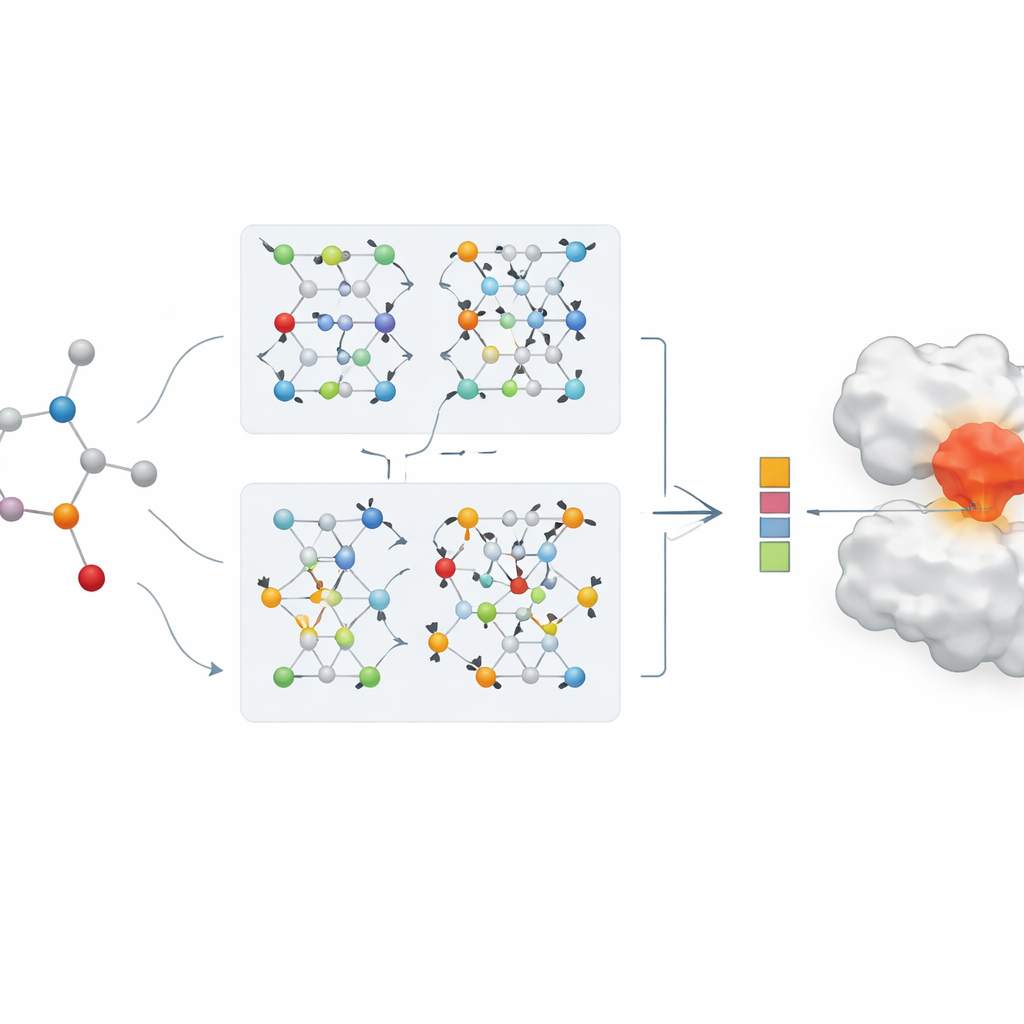
Från siffror till verkliga sjukdomsfall
För att visa verkligt värde tillämpar teamet sin modell i en studie om läkemedelsomplacering för COVID-19, med fokus på det huvudsakliga virala enzym som många behandlingar syftar till att inaktivera. AI:n rankar befintliga antivirala och närliggande läkemedel efter förutspådd bindningsstyrka till detta enzym. Flera välkända kandidater, inklusive remdesivir och andra proteashämmare, dyker upp högt på listan och stämmer överens med experimentella fynd som rapporterats av andra grupper. När modellens rankningar jämförs med traditionella datorbaserade dockningspoäng finns ett starkt överensstämmelse, och AI:n visar särskild styrka i att pusha sannolika träffar mot toppen. Detta tyder på att sådana modeller kan fungera som effektiva filter för att prioritera föreningar för experimentell testning vid snabbrörliga hälsokriser.
Vad detta betyder för framtidens läkemedelsupptäckt
Sammanfattningsvis visar studien att noggrant utformade grafbaserade AI-modeller kan ge mer informativa fingeravtryck av läkemedelsmolekyler och mer tillförlitliga förutsägelser av hur de interagerar med proteintargets. För icke-specialister är huvudbudskapet att smartare digitala representationer av kemi kan göra virtuell screening mer exakt och praktisk, särskilt när tid och resurser är begränsade. Även om metoden fortfarande står inför utmaningar — inklusive beräkningskostnad och känslighet för designval — utgör den ett viktigt steg mot AI-system som hjälper forskare att omplacera befintliga läkemedel och designa nya mer effektivt och säkert.
Citering: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Nyckelord: grafneuralnätverk, läkemedelsutveckling, prediktion av bindningsaffinitet, läkemedelsomplacering, beräkningsfarmakologi