Clear Sky Science · es
Una arquitectura de red neuronal gráfica de doble rama para la predicción de la afinidad de unión fármaco‑diana
Vías más rápidas hacia mejores medicamentos
Desarrollar un fármaco nuevo suele llevar más de una década y miles de millones de dólares, y solo una pequeña fracción de los candidatos llega a los pacientes. Este estudio explora cómo un nuevo tipo de inteligencia artificial, diseñada para entender las moléculas como redes de átomos conectados, puede acelerar la búsqueda de fármacos e incluso encontrar nuevos usos para medicamentos existentes. Al predecir con qué fuerza un fármaco se unirá a una proteína objetivo en el cuerpo, el enfoque pretende reducir miles de posibilidades a una lista corta manejable y de alta calidad.
Por qué importa predecir la “adhesión”
La mayoría de los fármacos modernos actúan uniéndose a proteínas específicas y alterando su comportamiento. La fuerza de esa unión, llamada afinidad de unión, está estrechamente ligada a si un fármaco será eficaz y a la dosis necesaria. Tradicionalmente, medir la afinidad de unión requiere experimentos de laboratorio laboriosos y grandes bibliotecas de compuestos químicos. Los autores se centran en reemplazar una parte de este proceso de prueba y error por computación: si un modelo informático puede predecir de manera fiable qué pares fármaco–proteína tienen más probabilidades de unirse con fuerza, los investigadores podrán ensayar muchas menos sustancias en el laboratorio, ahorrando tiempo y dinero y abriendo la puerta a nuevas ideas terapéuticas.
Ver las moléculas como mapas interconectados
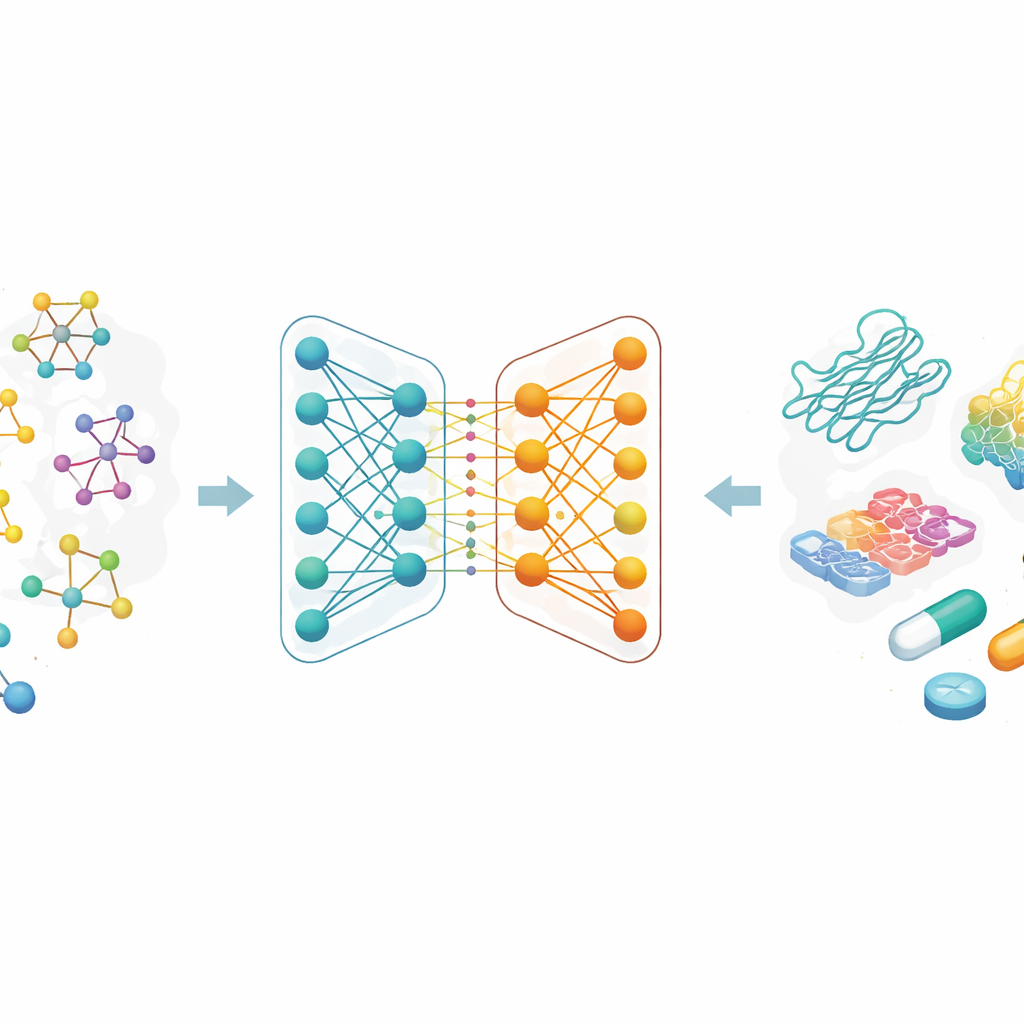
En lugar de describir las moléculas como cadenas de letras o largas listas de características manuales, los investigadores tratan cada fármaco como un grafo: los átomos son nodos y los enlaces químicos son las aristas entre ellos. Una familia de métodos de IA denominada redes neuronales gráficas es especialmente buena aprendiendo de estos mapas, porque transmite repetidamente información a lo largo de las conexiones, capturando tanto detalles locales como la estructura global. En este trabajo, cada fármaco parte de un código textual común en química, que luego se convierte en un grafo molecular. El equipo también utiliza codificaciones ricas para las proteínas objetivo, diseñadas para capturar patrones en las secuencias de aminoácidos y subestructuras con significado químico.
Dos ramas de IA que trabajan juntas
El núcleo del artículo es una nueva red neuronal gráfica de doble rama para la representación de fármacos. Una rama emplea una red de convolución sobre grafos, que mezcla la información de átomos vecinos de forma controlada. La otra utiliza un método llamado GraphSAGE, que aprende a resumir el vecindario de cada átomo a partir de los datos. Un módulo de "conocimiento saltante" (jumping knowledge) combina de forma flexible la información de distintas profundidades de la red, de modo que la representación final del fármaco refleja tanto patrones superficiales y locales como otros más profundos y globales. Esta codificación de doble rama se combina con un codificador de proteínas robusto, y las características combinadas fármaco–proteína se introducen en un módulo de predicción que produce una estimación de la fuerza de unión.
Qué tan bien funciona el modelo
Para probar su diseño, los autores lo comparan con 45 combinaciones distintas de codificadores de fármacos y proteínas existentes en dos colecciones de referencia ampliamente usadas: los conjuntos de datos Davis y KIBA, que documentan cómo interactúa una variedad de pequeñas moléculas con quinasas proteicas, una clase importante de dianas farmacológicas. Según varias medidas estándar de precisión y calidad de ordenamiento, el nuevo modelo de doble rama se sitúa de forma consistente en la cumbre o cerca de ella. En el conjunto Davis en particular, ofrece menor error de predicción y un mejor ordenamiento de los pares fármaco–diana que los métodos basados en grafos competidores, lo que sugiere que su visión más rica de la estructura molecular capta diferencias sutiles que modelos más simples pasan por alto. En el conjunto KIBA, más grande y ruidoso, las mejoras son menores pero aún robustas, lo que indica una buena capacidad de generalización.
De los números a casos reales de enfermedad
Para demostrar su valor en el mundo real, el equipo aplica su modelo a un estudio de reorientación de fármacos para COVID-19, centrado en la enzima viral principal que muchos tratamientos intentan inhibir. La IA clasifica fármacos antivirales existentes y otros relacionados según la predicción de su afinidad de unión a esa enzima. Varios candidatos bien conocidos, incluidos remdesivir y otros inhibidores de proteasas, aparecen cerca de la parte alta de la lista, coincidiendo con hallazgos experimentales reportados por otros grupos. Cuando se comparan las clasificaciones del modelo con las puntuaciones tradicionales de docking computacional, hay una fuerte concordancia, y la IA muestra especial capacidad para situar los posibles aciertos en lo alto. Esto sugiere que tales modelos podrían servir como filtros potentes para priorizar compuestos en pruebas experimentales durante crisis sanitarias de desarrollo rápido.
Qué implica esto para el futuro del descubrimiento de fármacos
En conjunto, el estudio muestra que una IA basada en grafos y bien diseñada puede proporcionar huellas digitales más informativas de las moléculas farmacológicas y predicciones más fiables de cómo interactúan con dianas proteicas. Para quienes no son especialistas, el mensaje clave es que representaciones digitales más inteligentes de la química pueden hacer que el cribado virtual sea más preciso y práctico, especialmente cuando el tiempo y los recursos son limitados. Aunque el enfoque aún enfrenta desafíos—incluidos el coste computacional y la sensibilidad a las decisiones de diseño—representa un paso significativo hacia sistemas de IA que ayuden a los científicos a reorientar fármacos existentes y diseñar otros nuevos de forma más eficiente y segura.
Cita: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Palabras clave: redes neuronales gráficas, descubrimiento de fármacos, predicción de afinidad de unión, repropósito de fármacos, farmacología computacional