Clear Sky Science · tr
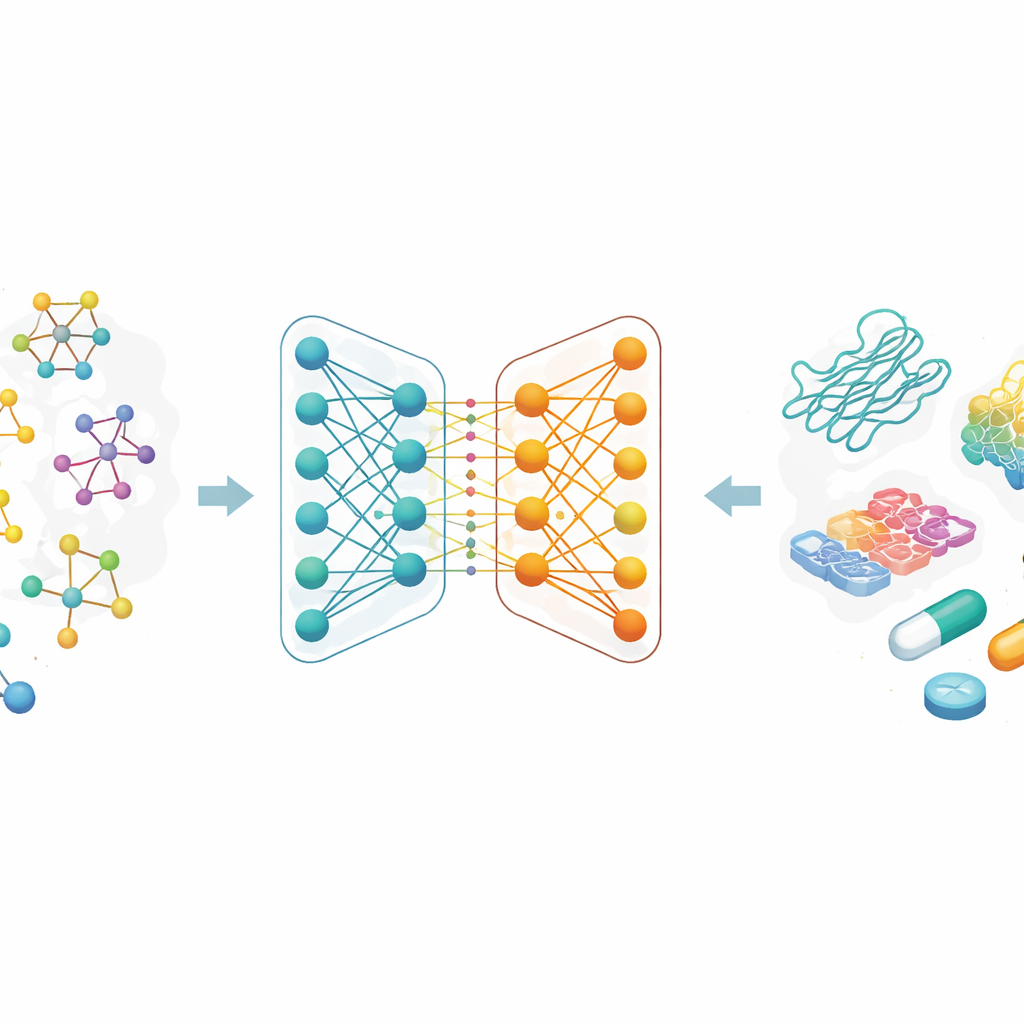
İlaç-hedef bağlanma afinitesi tahmini için çift dallı bir grafik sinir ağı mimarisi
Daha iyi ilaçlara giden daha hızlı yollar
Yeni bir ilaç geliştirmek genellikle on yılı aşkın sürenin ve milyarlarca doların harcanmasını gerektirir; adayların yalnızca küçük bir kısmı nihayetinde hastalara ulaşır. Bu çalışma, atomların bağlı olduğu ağlar olarak molekülleri anlamaya yönelik yeni bir yapay zekâ türünün ilaç arayışını nasıl hızlandırabileceğini ve mevcut ilaçlar için yeni kullanım alanları bulabileceğini inceliyor. Bir ilacın bedendeki bir protein hedefe ne kadar güçlü yapışacağını tahmin ederek yaklaşım, binlerce olasılığı yönetilebilir, yüksek kaliteli bir kısa listeye daraltmayı amaçlıyor.
Neden yapışkanlığı tahmin etmek önemli
Çoğu modern ilaç, belirli proteinlere tutunarak onların davranışını değiştirir. Bu tutunmanın gücü, bağlanma afinitesi olarak adlandırılır ve ilacın işe yarayıp yaramayacağı ve hangi dozda etkili olacağıyla yakından ilişkilidir. Geleneksel olarak bağlanma afinitesini ölçmek zahmetli laboratuvar deneyleri ve geniş kimyasal kütüphaneler gerektirir. Yazarlar, bu deneme-yanılma sürecinin bir bölümünü hesaplamayla değiştirmeye odaklanıyor: bir bilgisayar modeli hangi ilaç–protein çiftlerinin güçlü bağlanma olasılığı taşıdığını güvenilir biçimde tahmin edebilirse, araştırmacılar laboratuvarda çok daha az bileşiği test edebilir, hem zaman hem de maliyetten tasarruf sağlar ve yeni tedavi fikirlerinin önünü açar.
Molekülleri birbirine bağlı haritalar olarak görmek
Molekülleri harf dizileri veya elle tasarlanmış uzun özellik listeleri olarak tanımlamak yerine araştırmacılar her ilacı bir grafik olarak ele alıyor: atomlar düğümler, kimyasal bağlar ise bunlar arasındaki çizgiler. Graf sinir ağları adı verilen bir yapay zekâ yöntemleri ailesi, bu haritalardan öğrenmede özellikle iyidir, çünkü bağlantılar boyunca bilgiyi tekrarlı biçimde aktararak hem yerel ayrıntıları hem de küresel yapıyı yakalar. Bu çalışmada her ilaç, kimyada yaygın olarak kullanılan metin benzeri bir koddan başlatılır ve ardından moleküler bir grafiğe dönüştürülür. Ekip ayrıca amino asit dizilerindeki ve kimyasal açıdan anlamlı altyapılardaki desenleri yakalamayı amaçlayan zengin protein hedef kodlamaları da kullanır.
İki yapay zekâ dalı birlikte çalışıyor
Makalenin özü, ilaç temsili için yeni bir çift dallı grafik sinir ağıdır. Bir dal, komşu atomlardan gelen bilgiyi kontrollü biçimde harmanlayan bir grafik konvolüsyonel ağ kullanır. Diğer dal ise her atomun çevresini veriden özetlemeyi öğrenen GraphSAGE adlı bir yöntemi kullanır. Ardından bir “atlama bilgisi” (jumping knowledge) modülü, ağın farklı derinliklerindeki bilgileri esnek biçimde birleştirir; böylece nihai ilaç temsili hem sığ, yerel kalıpları hem de daha derin, daha küresel yapıları yansıtır. Bu çift dallı kodlama güçlü bir protein kodlayıcıyla eşleştirilir ve birleştirilmiş ilaç–protein özellikleri tahmin modülüne beslenerek tahmini bir bağlanma gücü çıktısı verir.
Modelin performansı ne kadar iyi
Tasarımı test etmek için yazarlar, iki yaygın kıyas koleksiyonunda—protein kinazları gibi önemli bir hedef sınıfıyla etkileşen çeşitli küçük moleküllerin kataloglandığı Davis ve KIBA veri setlerinde—mevcut ilaç ve protein kodlayıcılarının 45 farklı kombinasyonuyla karşılaştırma yapar. Bir dizi standart doğruluk ve sıralama kalitesi ölçütü boyunca yeni çift dallı model sürekli olarak en üstte veya yakınında yer alır. Özellikle Davis veri setinde, rakip grafik tabanlı yöntemlere kıyasla daha düşük tahmin hatası ve daha iyi ilaç–hedef çift sıralaması sunar; bu da daha basit modellerin kaçırdığı ince farkları yakalayan daha zengin bir moleküler yapı görüşüne işaret eder. Daha büyük ve daha gürültülü KIBA veri setinde iyileşmeler daha küçük ama hâlâ sağlamdır, bu da iyi genelleme yeteneğini gösterir.
Sayılaradan gerçek hastalık vakalarına
Gerçek dünya değerini göstermek için ekip, birçok tedavinin devre dışı bırakmayı hedeflediği ana viral enzim üzerine odaklanan COVID-19 için bir ilaç yeniden kullanım vaka çalışmasına modellerini uygular. Yapay zekâ mevcut antiviral ve ilgili ilaçları bu enzime tahmini bağlanma gücüne göre sıralar. Remdesivir ve bazı proteaz inhibitörleri de dahil olmak üzere birkaç iyi bilinen aday listenin üst sıralarında görünür; bu bulgular diğer grupların bildirdiği deneysel sonuçlarla uyum gösterir. Modelin sıralamaları geleneksel bilgisayar destekli docking (yerleştirme) skorlarıyla karşılaştırıldığında sağlam bir uyum vardır ve yapay zekâ muhtemel etkin bileşenleri üst sıralara itmedeki gücünü özellikle gösterir. Bu, böyle modellerin hızlı şekilde ilerleyen sağlık krizlerinde deneysel testler için bileşikleri önceliklendirecek güçlü filtreler olarak hizmet edebileceğini öne sürer.
Gelecekteki ilaç keşfi için bunun anlamı
Genel olarak çalışma, dikkatle tasarlanmış grafik tabanlı yapay zekânın ilaç moleküllerinin daha bilgilendirici parmak izlerini ve protein hedeflerle nasıl etkileştiklerine dair daha güvenilir tahminler sağlayabileceğini gösteriyor. Konunun uzmanı olmayanlar için temel mesaj, kimyanın daha akıllı dijital temsillerinin sanal taramayı daha doğru ve pratik hale getirebileceği, özellikle zaman ve kaynakların sınırlı olduğu durumlarda olduğudur. Yaklaşım hâlâ hesaplama maliyeti ve tasarım tercihlerine duyarlılık gibi zorluklarla karşılaşsa da, mevcut ilaçların yeniden kullanılmasına yardımcı olan ve yeni ilaçların daha verimli ve güvenli biçimde tasarlanmasını destekleyen yapay zekâ sistemlerine doğru anlamlı bir adımı temsil eder.
Atıf: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Anahtar kelimeler: graf sinir ağları, ilaç keşfi, bağlanma afinitesi tahmini, ilaç yeniden kullanımı, hesaplamalı farmakoloji