Clear Sky Science · ja
薬物-標的結合親和性予測のための二重分岐グラフニューラルネットワークアーキテクチャ
より良い薬への早い道筋
新薬の開発には通常10年以上、数十億ドルがかかり、候補のごく一部しか患者に届きません。本研究は、分子を結合した原子のネットワークとして理解するよう設計された新しい種類の人工知能が、どのようにして薬の探索を加速し、既存薬の新たな適応を見つけられるかを探ります。薬が体内のタンパク質標的にどれだけ強く結合するかを予測することで、数千の候補を扱いやすく高品質なショートリストに絞り込むことを目指しています。
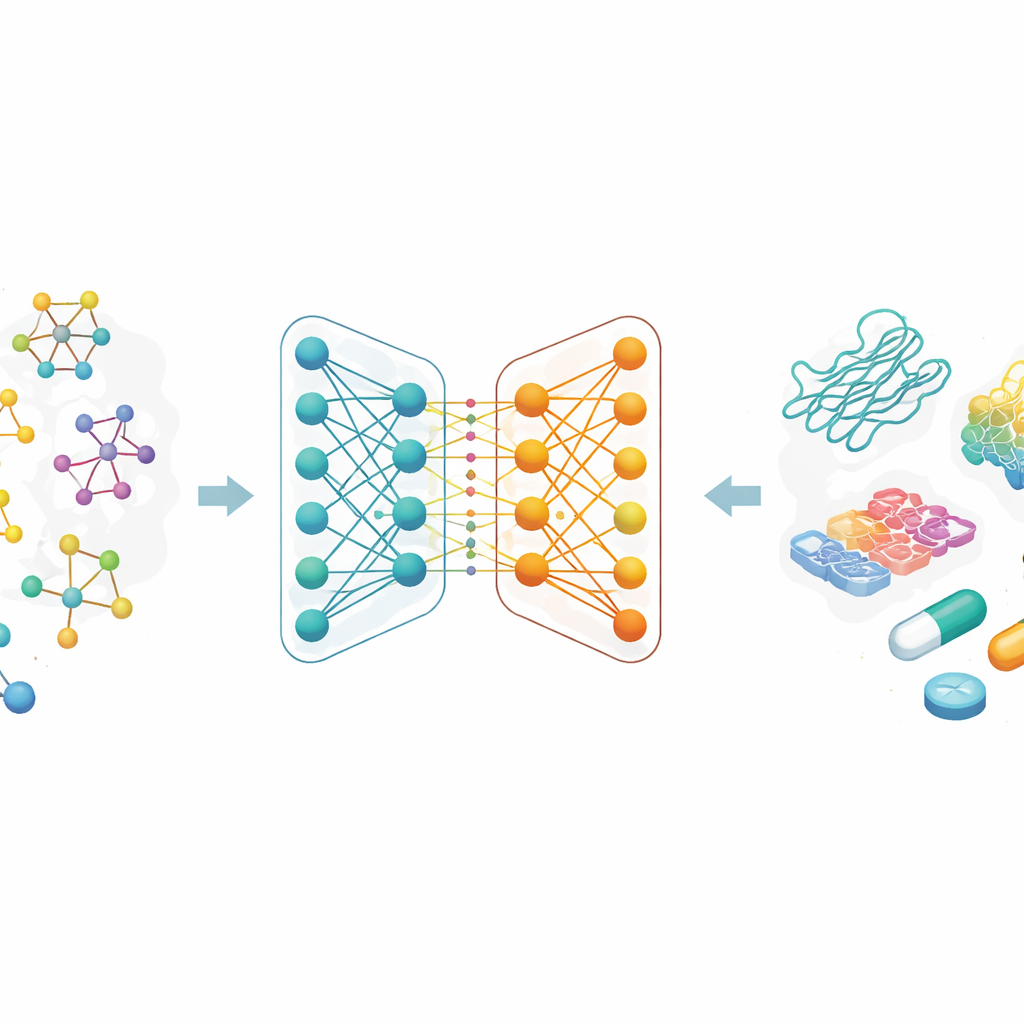
結合の“くっつきやすさ”を予測する意義
多くの現代薬は特定のタンパク質に結合してその挙動を変えることで作用します。その結合の強さ、すなわち結合親和性は、薬が効くかどうかや投与量と密接に関連しています。従来、結合親和性の計測は手間のかかる実験室での作業と大量の化合物ライブラリを必要とします。著者らは、この試行錯誤の一部を計算に置き換えることに注目しています。もしコンピュータモデルがどの薬–タンパク質の組み合わせが強く結合するかを信頼性高く予測できれば、研究者は実験で試す化合物を大幅に減らせ、時間と費用を節約しつつ新しい治療アイデアを生み出せます。
分子を相互接続された地図として捉える
分子を文字列や手作りの特徴の長い列として記述する代わりに、研究者たちは各薬物をグラフとして扱います:原子が点、化学結合がそれらを結ぶ線です。グラフニューラルネットワークと呼ばれる一群のAI手法は、これらの地図から学習するのに特に適しており、接続に沿って情報を反復的に伝播させることで局所的な詳細と大域的な構造の両方を捉えます。本研究では、各薬物は化学で一般に使われるテキスト風のコード(例:SMILESなど)から始められ、それが分子グラフに変換されます。著者らはまた、アミノ酸配列のパターンや化学的に意味のある部分構造を捉えるよう設計された豊かなエンコーディングをタンパク質標的にも用いています。
協働する二つのAI分岐
論文の中心は、薬物表現のための新しい二重分岐グラフニューラルネットワークです。一方の分岐は、隣接する原子からの情報を制御された方法で融合するグラフ畳み込みネットワークを用います。もう一方は、各原子の近傍をデータから要約する方法を学習するGraphSAGEという手法を使います。さらに「ジャンピング・ナレッジ」モジュールがネットワークの異なる深さからの情報を柔軟に組み合わせることで、最終的な薬物表現は浅い局所パターンと深い大域パターンの両方を反映します。この二重分岐エンコーディングは強力なタンパク質エンコーダーと組み合わされ、統合された薬物–タンパク質特徴が予測モジュールに入力され、推定される結合強度を出力します。
モデルの性能
設計を検証するために、著者らは既存の薬物およびタンパク質エンコーダーの45通りの組み合わせと、広く使われるベンチマークコレクションであるDavisおよびKIBAデータセット上で比較しています。これらはさまざまな小分子が薬物標的として重要なタンパク質キナーゼとどのように相互作用するかを記録したデータです。複数の標準的な精度およびランキング指標において、新しい二重分岐モデルは一貫して上位に位置します。特にDavisデータセットでは、競合するグラフベース手法よりも低い予測誤差とより良い薬物–標的ペアの順位付けを示し、単純なモデルでは見落としがちな微妙な違いを豊かに捉えていることを示唆します。より大きくノイジーなKIBAデータセットでは改善幅は小さいものの堅牢であり、良好な一般化性能を示しています。
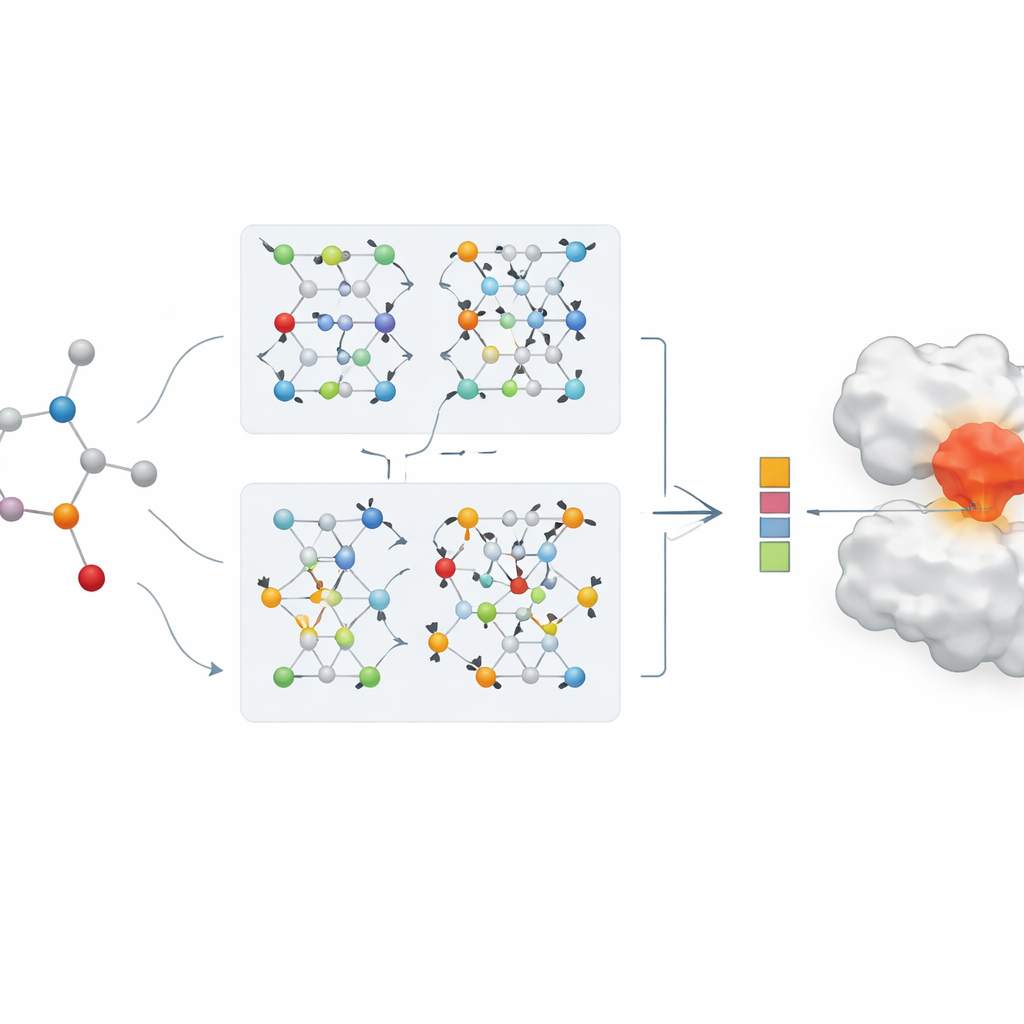
数値結果から実際の疾患ケースへ
実世界での有用性を示すため、チームはCOVID-19に対するドラッグリポジショニングのケーススタディにモデルを適用し、多くの治療が標的とする主要なウイルス酵素に注目しました。AIは既存の抗ウイルス薬や関連薬をこの酵素への結合強度の予測値で順位付けします。レムデシビルや他のプロテアーゼ阻害薬を含むいくつかの知られた候補がリストの上位に現れ、他グループの実験結果と一致しています。モデルのランキングを従来の計算ドッキングスコアと比較すると強い一致が見られ、特にヒット候補を上位に押し上げる点でAIは優れた性能を示しています。これは、こうしたモデルが迅速に動く健康危機の際に実験検査の優先順位付けを行う強力なフィルターになり得ることを示唆します。
今後の創薬への意味
総じて、本研究は注意深く設計されたグラフベースのAIが薬物分子のより情報量の多いフィンガープリントを提供し、タンパク質標的との相互作用のより信頼できる予測を可能にすることを示しています。非専門家にとっての主要なメッセージは、化学のデジタル表現を賢くすることで、特に時間や資源が限られる状況で仮想スクリーニングがより正確かつ実用的になる、という点です。計算コストや設計選択への感度といった課題は残るものの、このアプローチは既存薬のリポジショニングや新薬設計をより効率的かつ安全に支援するAIシステムへの意味ある一歩を示しています。
引用: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
キーワード: グラフニューラルネットワーク, 創薬, 結合親和性予測, ドラッグリポジショニング, 計算薬理学