Clear Sky Science · pl
Dwugałęziowa architektura grafowych sieci neuronowych do przewidywania powinowactwa wiązania lek–cel
Szybsze ścieżki do lepszych leków
Opracowanie nowego leku zwykle zajmuje ponad dekadę i kosztuje miliardy dolarów, przy czym tylko niewielka część kandydatów trafia do pacjentów. W tym badaniu analizuje się, jak nowy rodzaj sztucznej inteligencji — zaprojektowany do rozumienia cząsteczek jako sieci połączonych atomów — może przyspieszyć poszukiwania leków, a nawet znaleźć nowe zastosowania dla istniejących substancji. Poprzez przewidywanie, jak silnie lek będzie się wiązał z białkowym celem w organizmie, podejście ma na celu zawężenie tysięcy możliwości do praktycznej, wysokiej jakości krótkiej listy.
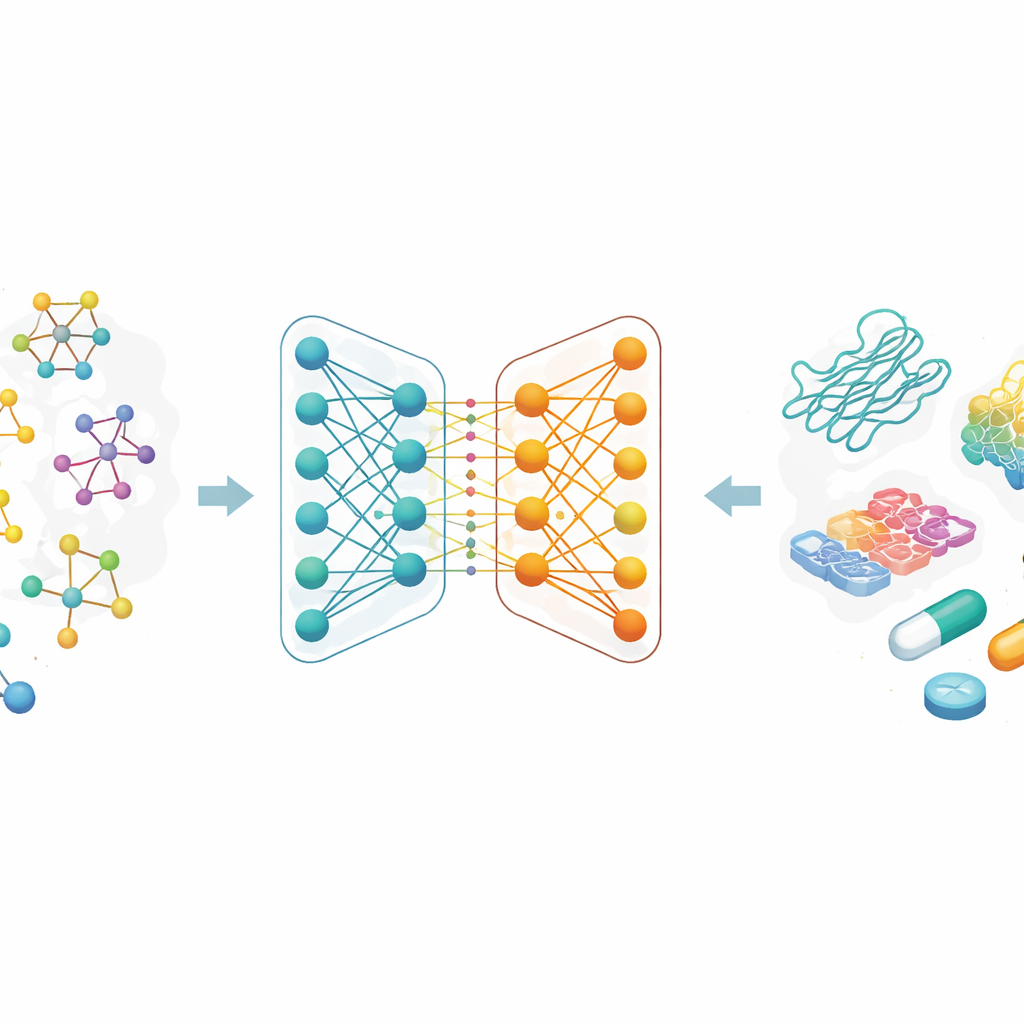
Dlaczego przewidywanie „przyczepności” ma znaczenie
Większość współczesnych leków działa przez przyłączanie się do określonych białek i modyfikowanie ich funkcji. Siła tego uchwytu, zwana powinowactwem wiązania, jest ściśle powiązana z tym, czy lek zadziała i w jakiej dawce. Tradycyjnie pomiar powinowactwa wymaga pracochłonnych eksperymentów laboratoryjnych i dużych bibliotek związków chemicznych. Autorzy koncentrują się na zastąpieniu części tego procesu metodą obliczeniową: jeśli model komputerowy może wiarygodnie przewidywać, które pary lek–białko będą się mocno wiązać, badacze mogą testować znacznie mniej związków w laboratorium, oszczędzając czas i pieniądze oraz otwierając drogę do nowych pomysłów terapeutycznych.
Postrzeganie cząsteczek jako powiązanych map
Zamiast opisywać cząsteczki jako ciągi liter lub długie listy ręcznie opracowanych cech, badacze traktują każdy lek jako graf: atomy są węzłami, a wiązania chemiczne — krawędziami między nimi. Rodzina metod AI zwanych grafowymi sieciami neuronowymi bardzo dobrze uczy się na takich mapach, ponieważ wielokrotnie przekazuje informacje wzdłuż połączeń, wychwytując zarówno lokalne szczegóły, jak i globalną strukturę. W tej pracy każdy lek zaczyna od powszechnie stosowanego tekstowego kodu chemicznego, który następnie jest konwertowany na graf molekularny. Zespół wykorzystuje też bogate kodowania dla białkowych celów, zaprojektowane tak, by uchwycić wzorce w sekwencjach aminokwasów oraz chemicznie istotne podstruktury.
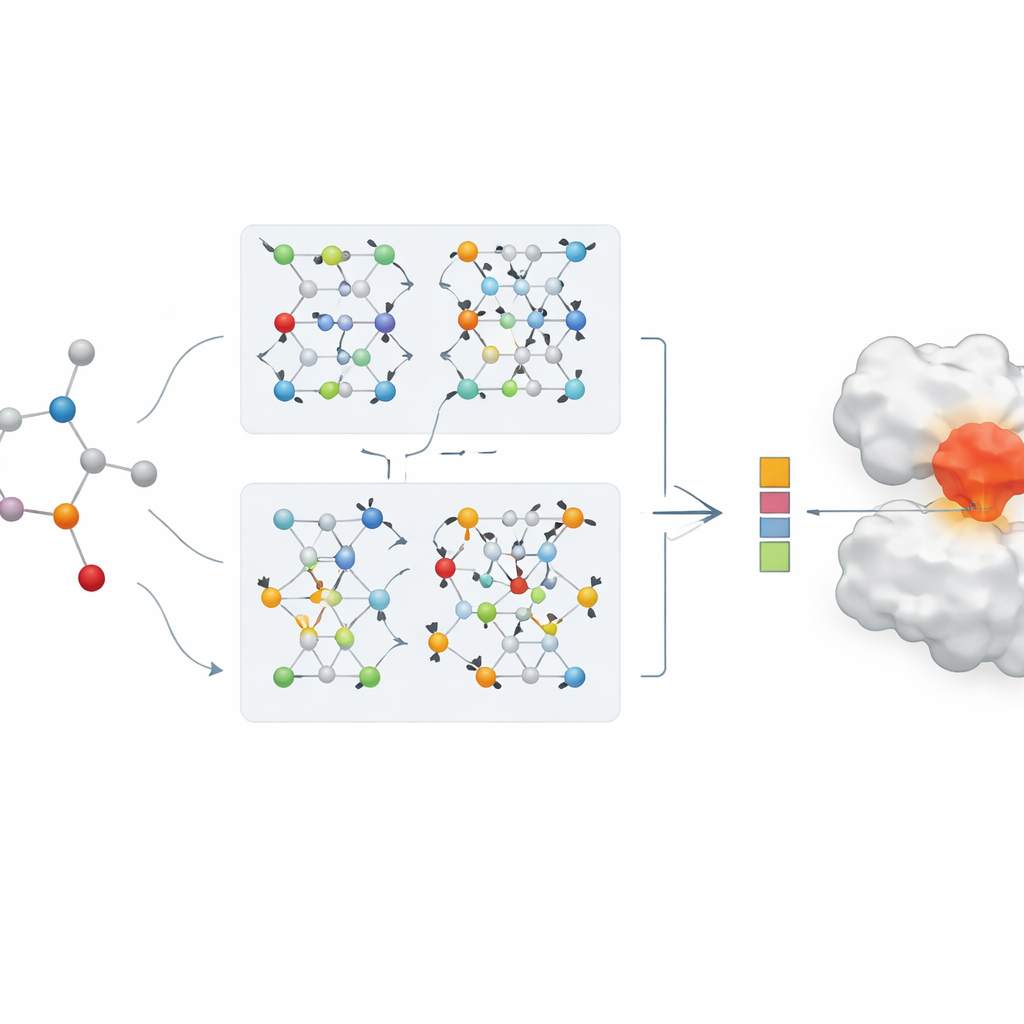
Dwie gałęzie AI współpracujące ze sobą
Rdzeń artykułu stanowi nowa dwugałęziowa grafowa sieć neuronowa do reprezentacji leków. Jedna gałąź używa grafowej sieci konwolucyjnej, która w kontrolowany sposób łączy informacje z sąsiednich atomów. Druga stosuje metodę nazwaną GraphSAGE, która uczy się, jak na podstawie danych streszczać otoczenie każdego atomu. Moduł „jumping knowledge” następnie elastycznie łączy informacje z różnych głębokości sieci, tak by ostateczna reprezentacja leku odzwierciedlała zarówno płytkie, lokalne wzorce, jak i głębsze, bardziej globalne. Ta dwugałęziowa enkodacja jest sparowana z mocnym enkoderem białek, a połączone cechy lek–białko są przekazywane do modułu predykcji, który zwraca szacowane natężenie wiązania.
Jak dobrze działa model
Aby przetestować swoje rozwiązanie, autorzy porównują je z 45 różnymi kombinacjami istniejących enkoderów leków i białek na dwóch szeroko stosowanych zbiorach testowych: Davis i KIBA, które katalogują interakcje różnych małych cząsteczek z kinazami białkowymi — istotną klasą celów lekowych. W kilku standardowych miarach dokładności i jakości rankingu nowy model dwugałęziowy konsekwentnie plasuje się na szczycie lub blisko niego. W szczególności na zestawie Davis osiąga niższy błąd predykcji i lepsze uporządkowanie par lek–cel niż konkurencyjne metody oparte na grafach, co sugeruje, że jego bogatsze spojrzenie na strukturę molekularną wychwytuje subtelne różnice, które prostsze modele pomijają. Na większym i bardziej hałaśliwym zbiorze KIBA poprawy są mniejsze, ale wciąż istotne, co wskazuje na dobrą uogólnialność.
Od liczb do rzeczywistych przypadków chorób
Aby pokazać wartość w praktyce, zespół zastosował swój model w studium przypadku repurposing’u leków na COVID-19, koncentrując się na głównym enzymie wirusa, który wiele terapii próbuje zablokować. AI klasyfikuje istniejące leki przeciwwirusowe i powiązane pod kątem przewidywanego powinowactwa do tego enzymu. Kilka dobrze znanych kandydatów, w tym remdesivir i inne inhibitory proteaz, znalazło się wysoko na liście, co zgadza się z wynikami eksperymentalnymi raportowanymi przez inne grupy. Porównanie rankingów modelu z tradycyjnymi wynikami dokowania komputerowego wykazuje silną zgodność, a system AI szczególnie dobrze wypada w wypychania prawdopodobnych trafień na szczyt listy. To sugeruje, że takie modele mogłyby służyć jako skuteczne filtry do priorytetyzacji związków do testów eksperymentalnych w szybkich kryzysach zdrowotnych.
Co to oznacza dla przyszłego odkrywania leków
Podsumowując, badanie pokazuje, że starannie zaprojektowana AI oparta na grafach może dostarczać bardziej informatywnych „odcisków palców” cząsteczek leków oraz bardziej wiarygodnych przewidywań ich interakcji z białkowymi celami. Dla osób niebędących specjalistami kluczowy przekaz jest taki, że inteligentniejsze cyfrowe reprezentacje chemii mogą uczynić wirtualne przesiewy dokładniejszymi i bardziej praktycznymi, zwłaszcza gdy czas i zasoby są ograniczone. Choć podejście wciąż stoi przed wyzwaniami — w tym kosztami obliczeniowymi i wrażliwością na wybory projektowe — stanowi znaczący krok w kierunku systemów AI, które pomagają naukowcom efektywniej i bezpieczniej repurposować istniejące leki oraz projektować nowe.
Cytowanie: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Słowa kluczowe: grafowe sieci neuronowe, odkrywanie leków, przewidywanie powinowactwa wiązania, repurposing leków, farmakologia obliczeniowa