Clear Sky Science · de
Eine zweigleisige Graph-Neural-Network-Architektur zur Vorhersage der Bindungsaffinität von Wirkstoff-Ziel-Interaktionen
Schnellere Wege zu besseren Medikamenten
Die Entwicklung eines neuen Medikaments dauert in der Regel mehr als ein Jahrzehnt und kostet Milliarden von Dollar, wobei nur ein kleiner Bruchteil der Kandidaten jemals bei Patienten ankommt. Diese Studie untersucht, wie eine neue Art künstlicher Intelligenz, die Moleküle als Netzwerke verbundener Atome versteht, die Suche nach Wirkstoffen beschleunigen und sogar neue Einsatzmöglichkeiten für vorhandene Medikamente aufdecken kann. Indem sie vorhersagt, wie stark ein Wirkstoff an ein Proteinziel im Körper bindet, zielt der Ansatz darauf ab, Tausende von Möglichkeiten auf eine handhabbare, qualitativ hochwertige Shortlist zu reduzieren.
Warum die Vorhersage der Haftstärke wichtig ist
Die meisten modernen Medikamente wirken, indem sie an spezifische Proteine andocken und deren Verhalten verändern. Die Stärke dieses Griffs, die Bindungsaffinität genannt wird, hängt eng damit zusammen, ob ein Wirkstoff wirkt und in welcher Dosis. Traditionell erfordert die Messung der Bindungsaffinität aufwändige Laborversuche und große Bibliotheken chemischer Verbindungen. Die Autoren konzentrieren sich darauf, einen Teil dieses Trial-and-Error-Prozesses durch Berechnungen zu ersetzen: Wenn ein Computermodell zuverlässig vorhersagen kann, welche Wirkstoff–Protein-Paare wahrscheinlich stark binden, können Forscher viel weniger Verbindungen im Labor testen, Zeit und Geld sparen und gleichzeitig neue Behandlungsideen ermöglichen.
Moleküle als vernetzte Landkarten sehen
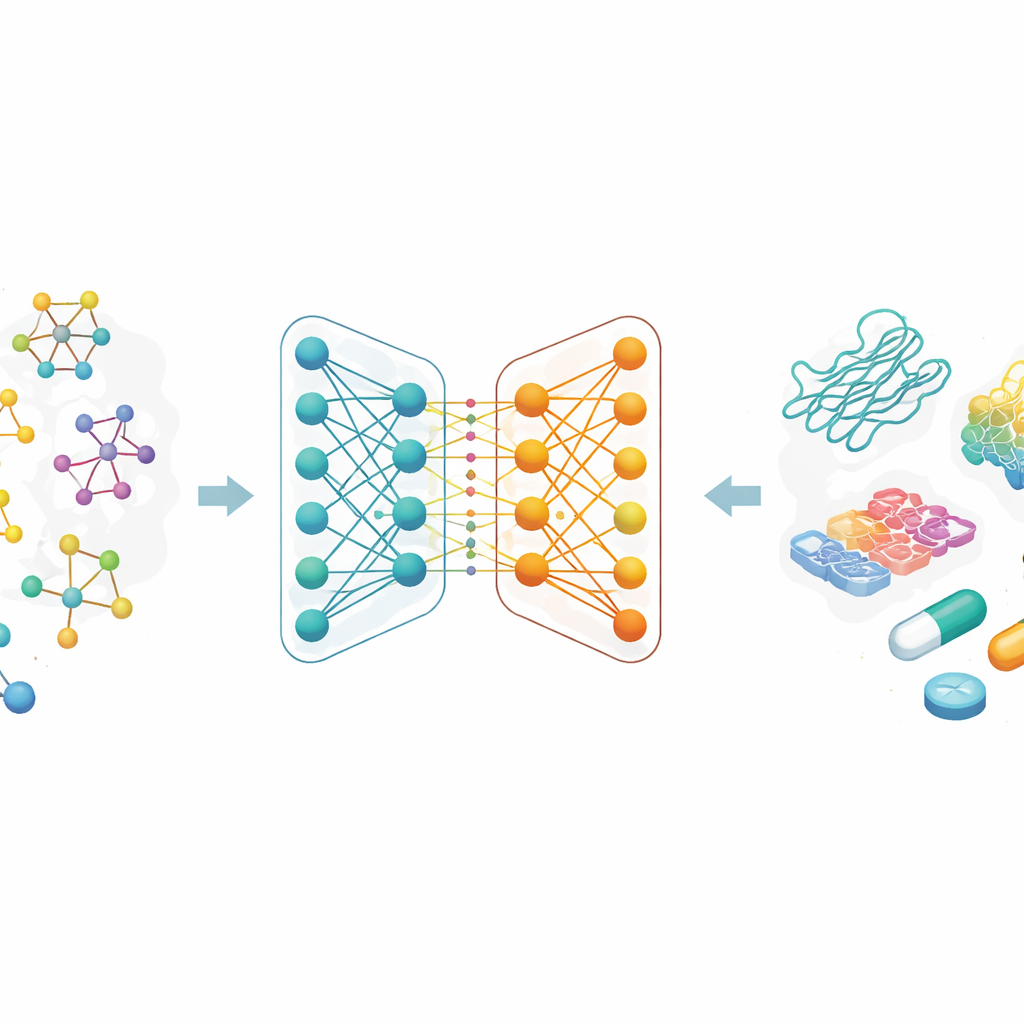
Anstatt Moleküle als Buchstabenketten oder lange Listen handgemachter Merkmale zu beschreiben, behandeln die Forschenden jeden Wirkstoff als Graph: Atome sind Punkte, und chemische Bindungen sind Linien zwischen ihnen. Eine Familie von KI-Methoden, die sogenannten Graph-Neural-Networks, eignet sich besonders gut zum Lernen aus solchen Karten, weil sie Informationen wiederholt entlang der Verbindungen weitergibt und so sowohl lokale Details als auch die globale Struktur erfasst. In dieser Arbeit beginnt jedes Wirkstoffmolekül mit einem verbreiteten, textähnlichen Code aus der Chemie, der dann in einen molekularen Graphen umgewandelt wird. Das Team verwendet außerdem reichhaltige Kodierungen für Proteinziele, die darauf ausgelegt sind, Muster in Aminosäuresequenzen und chemisch bedeutsame Substrukturen zu erfassen.
Zwei KI-Zweige, die zusammenarbeiten
Kern des Papiers ist ein neues zweigleisiges Graph-Neural-Network zur Darstellung von Wirkstoffen. Ein Zweig nutzt ein Graph-Convolutional-Network, das Informationen von benachbarten Atomen kontrolliert vermischt. Der andere verwendet eine Methode namens GraphSAGE, die lernt, wie man die Umgebung jedes Atoms aus Daten zusammenfasst. Ein „jumping knowledge“-Modul kombiniert dann flexibel Informationen aus verschiedenen Netzwerktiefen, sodass die endgültige Wirkstoffrepräsentation sowohl flache, lokale Muster als auch tiefere, globalere Merkmale widerspiegelt. Diese zweigleisige Kodierung wird mit einem leistungsfähigen Protein-Encoder gepaart, und die kombinierten Wirkstoff–Protein-Merkmale werden in ein Vorhersagemodul eingespeist, das eine geschätzte Bindungsstärke ausgibt.
Wie gut das Modell abschneidet
Um ihr Design zu testen, vergleichen die Autoren es mit 45 verschiedenen Kombinationen bestehender Wirkstoff- und Protein-Encoder an zwei weit verbreiteten Benchmark-Sammlungen: den Davis- und KIBA-Datensätzen, die katalogisieren, wie verschiedene kleine Moleküle mit Proteinkinasen interagieren, einer wichtigen Klasse von Arzneimittelzielen. Über mehrere Standardmaße für Genauigkeit und Ranking-Qualität liegt das neue zweigleisige Modell konsequent an oder nahe der Spitze. Insbesondere im Davis-Datensatz liefert es geringere Vorhersagefehler und eine bessere Reihenfolge von Wirkstoff–Ziel-Paaren als konkurrierende graphbasierte Methoden, was darauf hindeutet, dass seine reichere Sicht auf die Molekülstruktur subtile Unterschiede erfasst, die einfachere Modelle übersehen. Im größeren und stärker verrauschten KIBA-Datensatz sind die Verbesserungen kleiner, aber dennoch robust, was auf eine gute Generalisierung hinweist.
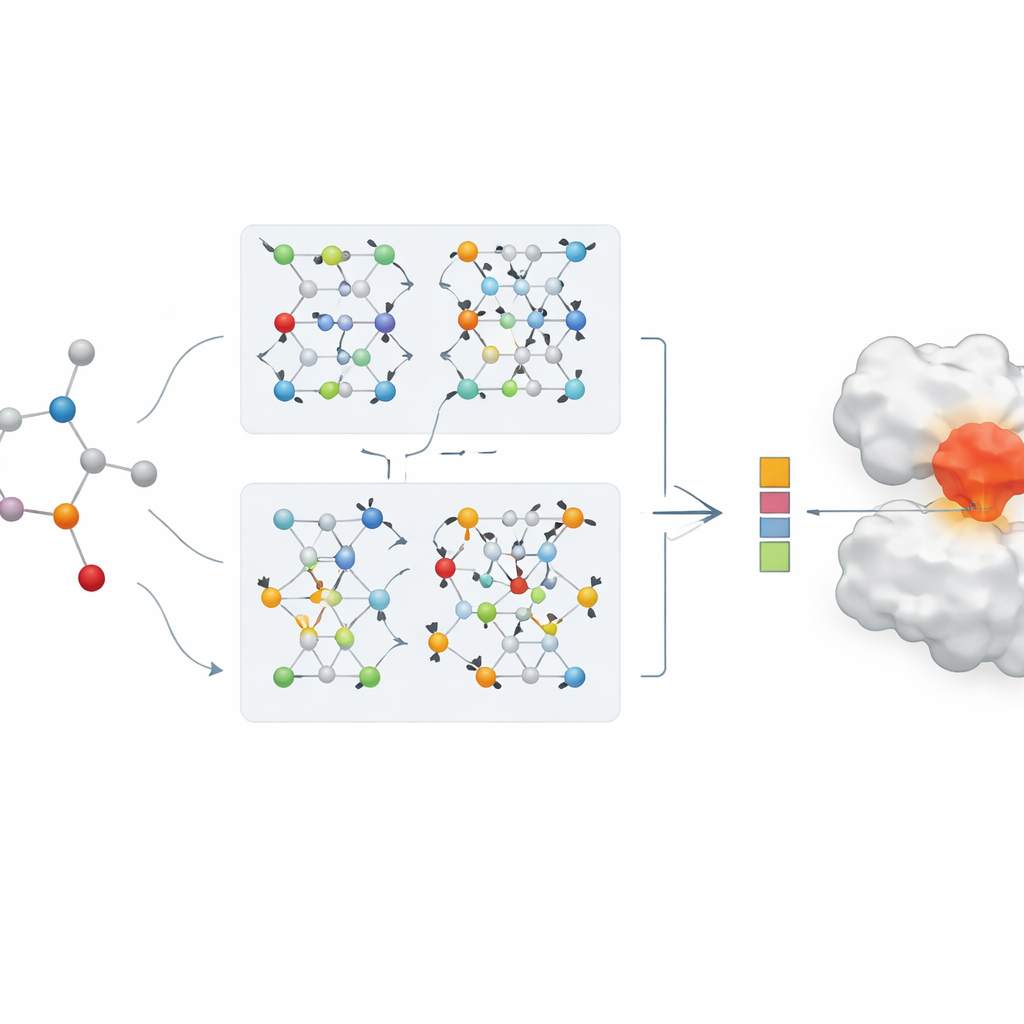
Von Zahlen zu realen Krankheitsfällen
Um den praktischen Nutzen zu demonstrieren, wendet das Team sein Modell auf eine Fallstudie zum Wirkstoff-Repurposing für COVID-19 an, mit Fokus auf das hauptsächliche virale Enzym, das viele Behandlungen zu deaktivieren versuchen. Die KI ordnet bestehende antivirale und verwandte Medikamente nach der vorhergesagten Bindungsstärke an dieses Enzym. Mehrere bekannte Kandidaten, darunter Remdesivir und andere Proteasehemmer, erscheinen weit oben auf der Liste und stimmen mit experimentellen Befunden anderer Gruppen überein. Wenn die Ranglisten des Modells mit traditionellen Docking-Scores verglichen werden, gibt es eine starke Übereinstimmung, und die KI zeigt besondere Stärke darin, wahrscheinliche Treffer nach oben zu schieben. Dies legt nahe, dass solche Modelle als leistungsfähige Filter dienen könnten, um Verbindungen während schnell verlaufender Gesundheitskrisen für experimentelle Tests zu priorisieren.
Was das für die zukünftige Wirkstoffforschung bedeutet
Insgesamt zeigt die Studie, dass sorgfältig entworfene graphbasierte KI aussagekräftigere Fingerabdrücke von Wirkstoffmolekülen und zuverlässigere Vorhersagen darüber liefern kann, wie sie mit Proteinzielen interagieren. Für Nicht-Spezialisten lautet die Kernbotschaft, dass intelligentere digitale Repräsentationen der Chemie das virtuelle Screening genauer und praktikabler machen können, besonders wenn Zeit und Ressourcen begrenzt sind. Obwohl der Ansatz weiterhin Herausforderungen gegenübersteht — darunter Rechenkosten und Sensitivität gegenüber Designentscheidungen — stellt er einen bedeutenden Schritt in Richtung KI-Systeme dar, die Wissenschaftlern helfen, vorhandene Medikamente neu zu nutzen und neue sicherer und effizienter zu entwerfen.
Zitation: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Schlüsselwörter: graph neural networks, Drug Discovery, Bindungsaffinitätsvorhersage, Drug Repurposing, computational pharmacology