Clear Sky Science · pt
Uma arquitetura de rede neural de grafos de dois ramos para predição da afinidade de ligação droga-alvo
Caminhos mais rápidos para medicamentos melhores
Desenvolver um novo medicamento normalmente leva mais de uma década e bilhões de dólares, com apenas uma pequena fração dos candidatos chegando aos pacientes. Este estudo explora como um novo tipo de inteligência artificial, projetada para entender moléculas como redes de átomos conectados, pode acelerar a busca por fármacos e até encontrar novos usos para medicamentos existentes. Ao prever quão fortemente uma droga se ligará a uma proteína-alvo no organismo, a abordagem pretende reduzir milhares de possibilidades a uma lista curta e de alta qualidade.
Por que prever a “adesão” é importante
A maioria dos medicamentos modernos atua ao se ligar a proteínas específicas e alterar seu comportamento. A força dessa ligação, chamada afinidade de ligação, está intimamente ligada a se um medicamento funcionará e em qual dose. Tradicionalmente, medir a afinidade de ligação exige experimentos laboratoriais demorados e grandes bibliotecas de compostos químicos. Os autores concentram-se em substituir parte desse processo de tentativa e erro por computação: se um modelo computacional puder prever com confiança quais pares droga–proteína provavelmente se ligarão fortemente, os pesquisadores podem testar muito menos compostos no laboratório, economizando tempo e dinheiro e abrindo caminho para novas ideias de tratamento.
Ver moléculas como mapas interconectados
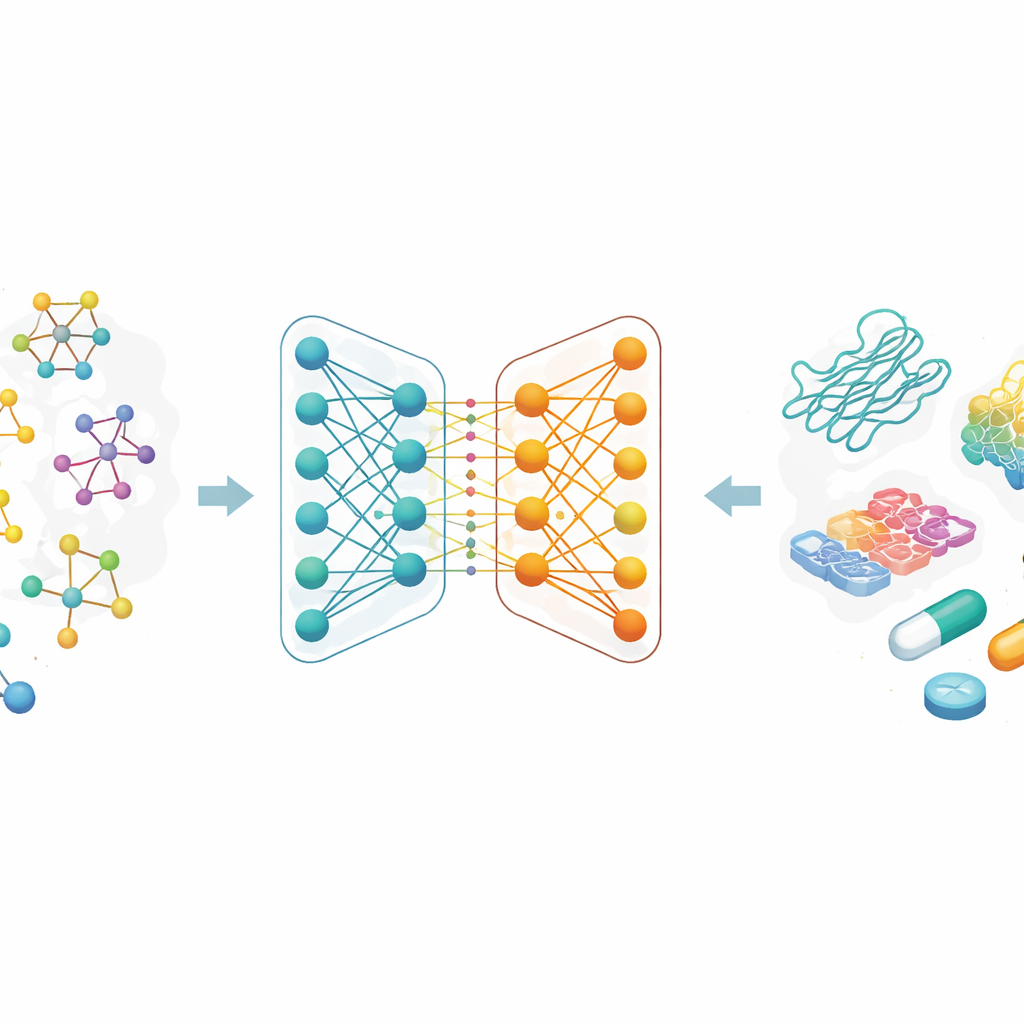
Em vez de descrever moléculas como sequências de letras ou longas listas de recursos elaborados manualmente, os pesquisadores tratam cada droga como um grafo: átomos são pontos e ligações químicas são linhas entre eles. Uma família de métodos de IA chamada redes neurais de grafos é particularmente boa em aprender a partir desses mapas, porque transmite informação repetidamente ao longo das conexões, capturando tanto detalhes locais quanto a estrutura global. Neste trabalho, cada droga parte de um código textual comum usado em química, que é então convertido em um grafo molecular. A equipe também usa codificações ricas para os alvos proteicos, projetadas para capturar padrões em sequências de aminoácidos e subestruturas quimicamente significativas.
Dois ramos de IA trabalhando juntos
O cerne do artigo é uma nova rede neural de grafos de dois ramos para representação de drogas. Um ramo usa uma rede convolucional de grafos, que mistura informações dos átomos vizinhos de maneira controlada. O outro usa um método chamado GraphSAGE, que aprende a resumir o entorno de cada átomo a partir dos dados. Um módulo de “conhecimento saltante” (jumping knowledge) então combina de forma flexível informações de diferentes profundidades da rede, de modo que a representação final da droga reflita padrões rasos e locais e também padrões mais profundos e globais. Essa codificação de dois ramos é emparelhada com um codificador forte para proteínas, e as características combinadas droga–proteína são alimentadas em um módulo de predição que fornece uma estimativa da força de ligação.
Quão bem o modelo se sai
Para testar seu projeto, os autores o comparam com 45 combinações diferentes de codificadores de drogas e proteínas existentes em duas coleções de referência amplamente utilizadas: os conjuntos de dados Davis e KIBA, que catalogam como uma variedade de pequenas moléculas interage com quinases proteicas, uma classe importante de alvos farmacológicos. Em várias medidas padrão de precisão e qualidade de ordenação, o novo modelo de dois ramos consistentemente fica no topo ou próximo dele. No conjunto Davis em particular, ele entrega menor erro de predição e melhor ordenação dos pares droga–alvo do que métodos concorrentes baseados em grafos, sugerindo que sua visão mais rica da estrutura molecular captura diferenças sutis que modelos mais simples deixam passar. No conjunto maior e mais ruidoso KIBA, as melhorias são menores, mas ainda robustas, indicando boa generalização.
Dos números a casos reais de doença
Para demonstrar valor no mundo real, a equipe aplica seu modelo a um estudo de caso de reposicionamento de medicamentos para COVID-19, focando na principal enzima viral que muitos tratamentos tentam desativar. A IA classifica antivirais existentes e drogas relacionadas pela força de ligação prevista a essa enzima. Vários candidatos bem conhecidos, incluindo remdesivir e outros inibidores de protease, aparecem perto do topo da lista, em concordância com achados experimentais relatados por outros grupos. Quando as classificações do modelo são comparadas com pontuações tradicionais de acoplamento computacional (docking), há forte acordo, e a IA mostra força particular em impulsionar os prováveis acertos para o topo. Isso sugere que tais modelos podem servir como filtros poderosos para priorizar compostos para testes experimentais durante crises de saúde de rápida evolução.
O que isso significa para a descoberta futura de fármacos
No geral, o estudo mostra que IA baseada em grafos e cuidadosamente projetada pode fornecer impressões digitais mais informativas de moléculas de droga e previsões mais confiáveis de como elas interagem com alvos proteicos. Para não especialistas, a mensagem-chave é que representações digitais mais inteligentes da química podem tornar o triagem virtual mais precisa e prática, especialmente quando tempo e recursos são limitados. Embora a abordagem ainda enfrente desafios — incluindo custo computacional e sensibilidade a escolhas de projeto — ela representa um passo significativo em direção a sistemas de IA que ajudam cientistas a reposicionar medicamentos existentes e projetar novos de forma mais eficiente e segura.
Citação: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Palavras-chave: redes neurais de grafos, descoberta de fármacos, predição de afinidade de ligação, reposicionamento de medicamentos, farmacologia computacional