Clear Sky Science · it
Una architettura di rete neurale a grafi a doppio ramo per la predizione dell’affinità di legame farmaco‑bersaglio
Percorsi più rapidi verso farmaci migliori
Sviluppare un nuovo farmaco generalmente richiede più di un decennio e miliardi di dollari, con solo una piccola frazione dei candidati che arriva mai ai pazienti. Questo studio esplora come un nuovo tipo di intelligenza artificiale, progettata per interpretare le molecole come reti di atomi connessi, possa accelerare la ricerca di farmaci e persino individuare nuovi usi per medicinali esistenti. Predicendo quanto saldamente un farmaco si legherà a una proteina bersaglio nel corpo, l’approccio mira a ridurre migliaia di possibilità a una rosa gestibile e di alta qualità.
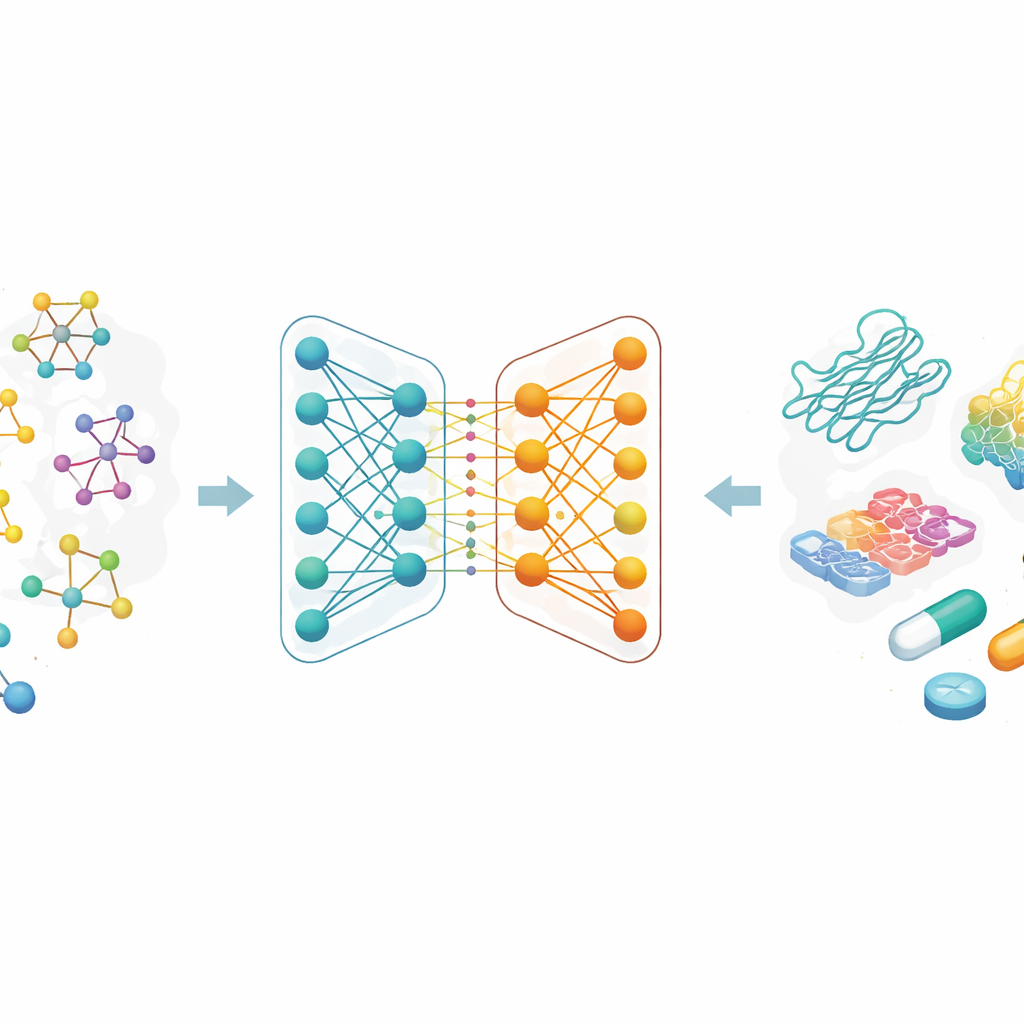
Perché la predizione della “aderenza” è importante
La maggior parte dei farmaci moderni agisce attaccandosi a proteine specifiche e modificandone il comportamento. La forza di quella presa, chiamata affinità di legame, è strettamente correlata al fatto che un farmaco funzioni e a quale dose. Tradizionalmente, misurare l’affinità di legame richiede esperimenti di laboratorio laboriosi e grandi librerie di composti chimici. Gli autori si concentrano sulla sostituzione di una parte di questo processo di prova‑ed‑errore con il calcolo: se un modello computazionale può prevedere in modo affidabile quali coppie farmaco–proteina sono probabilmente ad alta affinità, i ricercatori possono testare molto meno in laboratorio, risparmiando tempo e denaro e aprendo la strada a nuove idee terapeutiche.
Vedere le molecole come mappe interconnesse
Invece di descrivere le molecole come stringhe di lettere o lunghe liste di caratteristiche progettate a mano, i ricercatori trattano ogni farmaco come un grafo: gli atomi sono punti e i legami chimici sono linee tra di essi. Una famiglia di metodi di IA chiamata reti neurali a grafi è particolarmente adatta a imparare da queste mappe, perché trasmette ripetutamente informazioni lungo le connessioni, catturando sia dettagli locali sia la struttura globale. In questo lavoro, ogni farmaco parte da un codice simile a testo comunemente usato in chimica, che viene poi convertito in un grafo molecolare. Il gruppo utilizza inoltre codifiche ricche per i bersagli proteici, progettate per catturare pattern nelle sequenze di amminoacidi e sotto‑strutture chimicamente rilevanti.
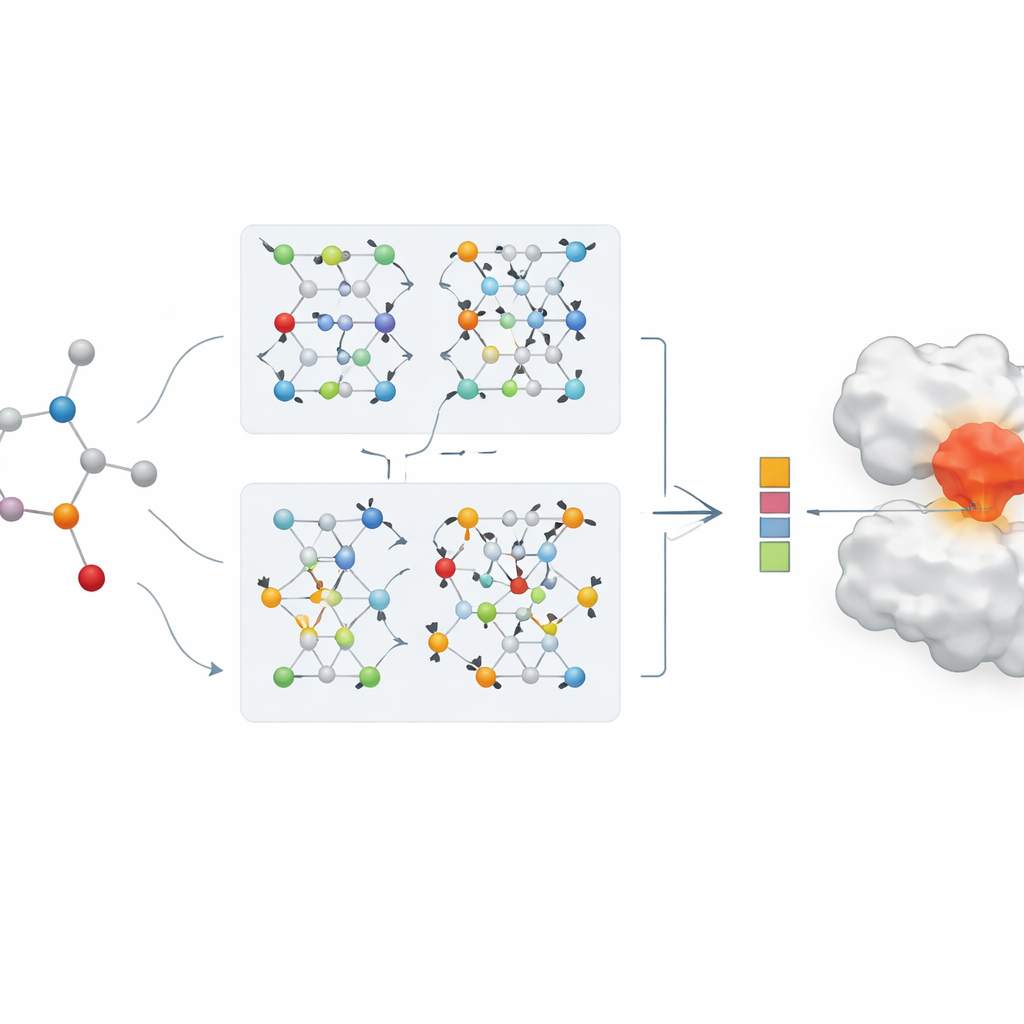
Due rami di IA che lavorano insieme
Il nucleo dell’articolo è una nuova rete neurale a grafi a doppio ramo per la rappresentazione dei farmaci. Un ramo utilizza una rete convoluzionale su grafi, che fonde le informazioni dagli atomi vicini in modo controllato. L’altro impiega un metodo chiamato GraphSAGE, che impara a sintetizzare il vicinato di ciascun atomo a partire dai dati. Un modulo di “jumping knowledge” combina quindi in modo flessibile informazioni provenienti da diverse profondità della rete, in modo che la rappresentazione finale del farmaco rifletta sia pattern superficiali e locali sia caratteristiche più profonde e globali. Questa codifica a doppio ramo è affiancata da un solido codificatore per le proteine, e le caratteristiche combinate farmaco–proteina vengono immesse in un modulo di predizione che restituisce una stima della forza di legame.
Quanto bene si comporta il modello
Per testare il loro progetto, gli autori lo confrontano con 45 diverse combinazioni di codificatori per farmaci e proteine esistenti su due collezioni di benchmark ampiamente usate: i dataset Davis e KIBA, che catalogano come una varietà di piccole molecole interagisce con le proteine chinasi, una classe importante di bersagli farmaceutici. Su varie misure standard di accuratezza e qualità di ranking, il nuovo modello a doppio ramo si posiziona costantemente al vertice o vicino ad esso. Sul dataset Davis in particolare, fornisce un errore di predizione inferiore e un ordinamento migliore delle coppie farmaco–bersaglio rispetto ai metodi concorrenti basati su grafi, suggerendo che la sua visione più ricca della struttura molecolare cattura differenze sottili che modelli più semplici perdono. Sul più grande e rumoroso dataset KIBA, i miglioramenti sono più contenuti ma ancora robusti, indicando una buona capacità di generalizzazione.
Dai numeri ai casi reali di malattia
Per dimostrare il valore nel mondo reale, il team applica il proprio modello a uno studio di caso sul riutilizzo di farmaci per il COVID‑19, concentrandosi sull’enzima virale principale che molti trattamenti mirano a disabilitare. L’IA classifica farmaci antivirali esistenti e composti correlati in base all’affinità di legame prevista verso questo enzima. Diversi candidati ben noti, tra cui remdesivir e altri inibitori della proteasi, compaiono vicino alla cima della lista, in linea con risultati sperimentali riportati da altri gruppi. Quando i ranking del modello sono confrontati con i punteggi tradizionali di docking computazionale, emerge una forte concordanza, e l’IA mostra particolare efficacia nel portare i probabili “hit” verso l’alto della graduatoria. Ciò suggerisce che tali modelli potrebbero fungere da filtri potenti per prioritizzare i composti per i test sperimentali durante crisi sanitarie in rapida evoluzione.
Cosa significa per il futuro della scoperta di farmaci
Nel complesso, lo studio mostra che un’IA basata su grafi progettata con cura può fornire impronte digitali più informative delle molecole farmaceutiche e previsioni più affidabili di come interagiscono con i bersagli proteici. Per i non specialisti, il messaggio chiave è che rappresentazioni digitali più intelligenti della chimica possono rendere lo screening virtuale più accurato e praticabile, specialmente quando tempo e risorse sono limitati. Pur affrontando ancora sfide — tra cui i costi computazionali e la sensibilità alle scelte progettuali — rappresenta un passo significativo verso sistemi di IA che aiutino gli scienziati a riutilizzare farmaci esistenti e a progettare nuovi composti in modo più efficiente e sicuro.
Citazione: Abbas, K., Hao, C., Dong, S. et al. A dual-branch graph neural network architecture for drug-target binding affinity prediction. Sci Rep 16, 13864 (2026). https://doi.org/10.1038/s41598-026-43782-4
Parole chiave: reti neurali a grafi, scoperta di farmaci, predizione dell’affinità di legame, riutilizzo di farmaci, farmacologia computazionale