Clear Sky Science · zh
PARK19 截短突变体 Dnajc6 导致溶酶体缺陷性上调病理性 α-突触核蛋白并在 PARK19 敲入小鼠中引起黑质多巴胺能细胞的神经退行性变
这项小鼠研究为何与帕金森病相关
帕金森病通过缓慢消灭一小群产生化学递质多巴胺的脑细胞,剥夺人们的运动能力。大多数病例由年龄和环境共同作用引起,但少数病例由单个基因缺陷造成,为理解疾病机制提供了有力线索。本研究使用一种经特殊工程改造的小鼠研究其中一个基因 DNAJC6,揭示了细胞内部微小改变如何削弱其废物处理系统,使其被一种称为 α-突触核蛋白的粘性蛋白负荷过重,并最终导致神经元丧失和类帕金森症状。
神经细胞中的有缺陷辅助手蛋白
基因 DNAJC6 编码一种几乎专门存在于神经元中的蛋白质,它帮助管理网格蛋白(clathrin),这是一种如外衣般的分子,参与塑形和回收细胞膜。在若干携带早发型帕金森病(称为 PARK19)的家系中,DNAJC6 的两份拷贝都存在破坏性变化,使蛋白的关键末端被截断。为了模拟这种情况,研究人员使用 CRISPR 基因编辑制造了小鼠的等效突变(小鼠中称为 Dnajc6),使其在对应位置提前终止。纯合的“Q787X”小鼠只产生截短的蛋白形式,而杂合小鼠则携带一份正常和一份突变拷贝,类似人类家系中的健康携带者。
从基因缺陷到运动障碍
研究团队随访小鼠随年龄的变化并测试其运动能力。在五个月大时,各组小鼠行为正常。然而到了九个月,携带两份突变拷贝的小鼠出现了典型的帕金森样运动问题:移动变慢、活动距离缩短,并且下竿测试所需时间增加,这类似患者的运动减少和缓慢。当科学家检查其大脑时,发现黑质中产生多巴胺的神经元显著丧失,纹状体的神经末梢变薄。这些易受损的神经元还含有磷酸化的 α-突触核蛋白团块,类似人类帕金森病中的路易体病理。只有一份突变拷贝的杂合小鼠没有这些变化。
当细胞回收系统崩溃
为了解事件链,作者检查了细胞的回收中心——溶酶体。在突变小鼠的多巴胺能神经元中,网格蛋白重链水平降低,预计会扰乱一种称为自噬性溶酶体再生(autophagic lysosome reformation)的过程——即细胞补充溶酶体库的方式。果然,溶酶体数量下降,且自噬受损的标志上升。关键的是,一种重要的溶酶体酶——半胱天冬酶 D(cathepsin D)降低,而 α-突触核蛋白及其更具毒性的磷酸化和寡聚形式积累并变得对降解有抵抗性。这种溶酶体失效具有选择性:邻近区域的抑制性神经元并未表现出相同缺陷,突显了黑质多巴胺细胞的特殊脆弱性。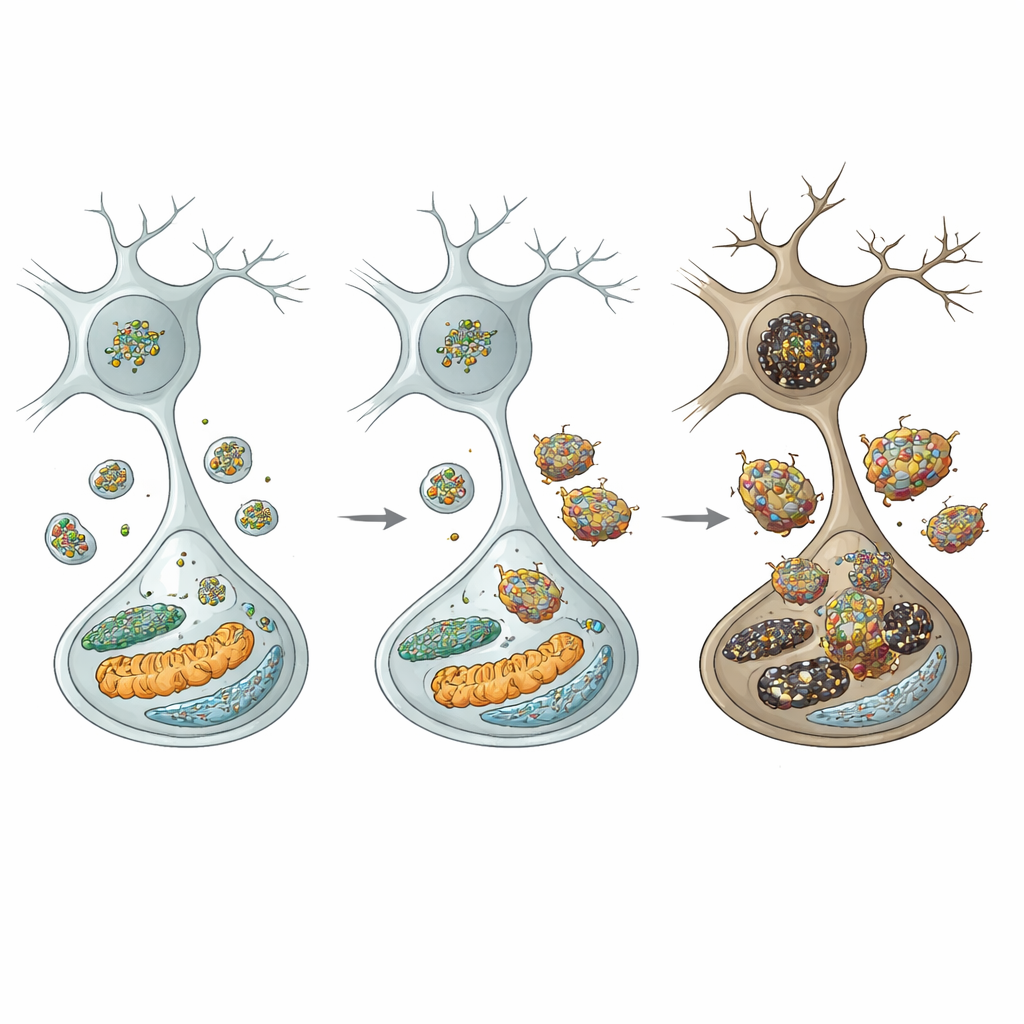
神经元内外的扩散性损伤
过量的 α-突触核蛋白并未停留在原位。它在两个关键的细胞内部结构——内质网和线粒体上积聚,触发应激和损伤。在内质网中,它激活了未折叠蛋白应激反应和促死信号。在线粒体中,它降低了能量产生复合体 I 的活性,增加活性氧(ROS),并促使细胞色素 c 释放,进而激活所谓的凋亡酶(半胱天冬酶)。同时,寡聚的 α-突触核蛋白可能从神经元外泄,激活附近的胶质细胞——脑内的免疫哨兵。这些小胶质细胞转向促炎状态,启动 NLRP3 炎性小体复合体,并释放如 IL-1β、IL-18 和 TNF-α 等炎性分子。这些信号反过来又在多巴胺神经元内激活额外的死亡通路,包括由 JNK 驱动的途径和坏死样程序性细胞死亡(necroptosis),形成互相强化的损伤恶性循环。
能恢复细胞清理系统的药物
研究者接着探索提高溶酶体生成能否打断这一级联反应。他们用雷帕霉素处理年轻的突变小鼠,雷帕霉素间接释放一个名为 TFEB 的主开关,使其进入细胞核并启动溶酶体构建基因,同时也刺激自噬。四个月后,雷帕霉素处理的突变小鼠在黑质中显示出溶酶体标志物和半胱天冬酶 D 水平恢复,且 α-突触核蛋白积累显著减少。值得注意的是,这些小鼠在黑质和邻近的腹侧被盖区的多巴胺神经元丧失方面大体受到保护,运动表现也朝正常方向改善。通过在细胞废物处理网络的上游作用,雷帕霉素有效减弱了蛋白积累、细胞器应激、炎症和细胞死亡的下游级联反应。
对理解和治疗帕金森病的意义
简言之,这项研究展示了神经元特异性辅助手蛋白的单一遗传缺陷如何引发一连串反应:溶酶体减少、半胱天冬酶 D 活性减弱、有毒 α-突触核蛋白水平上升、关键细胞隔室受损、炎性过度反应,最终导致支持帕金森症状的多巴胺神经元丧失。尽管这种特定突变罕见,但相同的主题——溶酶体缺陷、蛋白清除不良和 α-突触核蛋白过载——也出现在更常见的非家族性帕金森病中。因此,这项工作强化了这样的观点:通过恢复大脑清理系统,例如激活 TFEB 或增强自噬的药物(如雷帕霉素或更安全的后续药物),可能是减缓或预防与帕金森相关神经退行性变的有前景策略。
引用: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
关键词: 帕金森病, 溶酶体, α-突触核蛋白, 多巴胺能神经元, 雷帕霉素