Clear Sky Science · en
PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice
Why this mouse study matters for Parkinson’s disease
Parkinson’s disease robs people of movement by slowly killing a small group of brain cells that make the chemical messenger dopamine. Most cases arise from a mix of age and environment, but a minority are caused by single-gene faults that act as powerful clues to the disease process. This study examines one such gene, DNAJC6, using a specially engineered mouse to reveal how a tiny change deep inside brain cells can cripple their waste‑disposal systems, overload them with a sticky protein called alpha‑synuclein, and ultimately lead to neuron loss and Parkinson’s‑like symptoms. 
A faulty helper protein in brain cells
The gene DNAJC6 makes a protein found almost exclusively in neurons, where it helps manage clathrin, a coat-like molecule that shapes and recycles cell membranes. In several families with an early‑onset form of Parkinson’s disease known as PARK19, both copies of DNAJC6 carry damaging changes that chop off a critical end of the protein. To mimic this situation, the researchers used CRISPR gene editing to create mice whose version of the gene (called Dnajc6 in mice) stops early at an equivalent position. These homozygous “Q787X” mice produce only the shortened form of the protein, while heterozygous mice carry one normal and one mutant copy, similar to healthy carriers in human families.
From gene defect to movement problems
The team followed the mice as they aged and tested their movement. At five months, all groups behaved normally. By nine months, however, mice with two mutant copies developed classic Parkinson‑like motor problems: they moved more slowly, covered shorter distances, and needed extra time to descend a testing pole, mirroring hypokinesia and bradykinesia in patients. When the scientists examined their brains, they saw a marked loss of dopamine‑producing neurons in the substantia nigra and thinning of their nerve endings in the striatum. These vulnerable neurons also contained clumps of phosphorylated alpha‑synuclein, akin to the Lewy body pathology seen in human Parkinson’s disease. Heterozygous mice, with only one mutant copy, showed none of these changes.
When cellular recycling breaks down
To understand the chain of events, the authors looked at the cell’s recycling centers: lysosomes. In dopamine neurons from mutant mice, levels of clathrin heavy chain were reduced, which is expected to disrupt a process called autophagic lysosome reformation—the way cells replenish their lysosome pool. Indeed, the number of lysosomes fell, and markers of impaired cellular “self‑eating” (autophagy) rose. Crucially, a key lysosomal enzyme, cathepsin D, dropped, while alpha‑synuclein and its more toxic phosphorylated and oligomeric forms built up and even became resistant to digestion. This lysosomal failure was selective: neighboring inhibitory neurons in other regions did not show the same deficits, highlighting the particular vulnerability of substantia nigra dopamine cells. 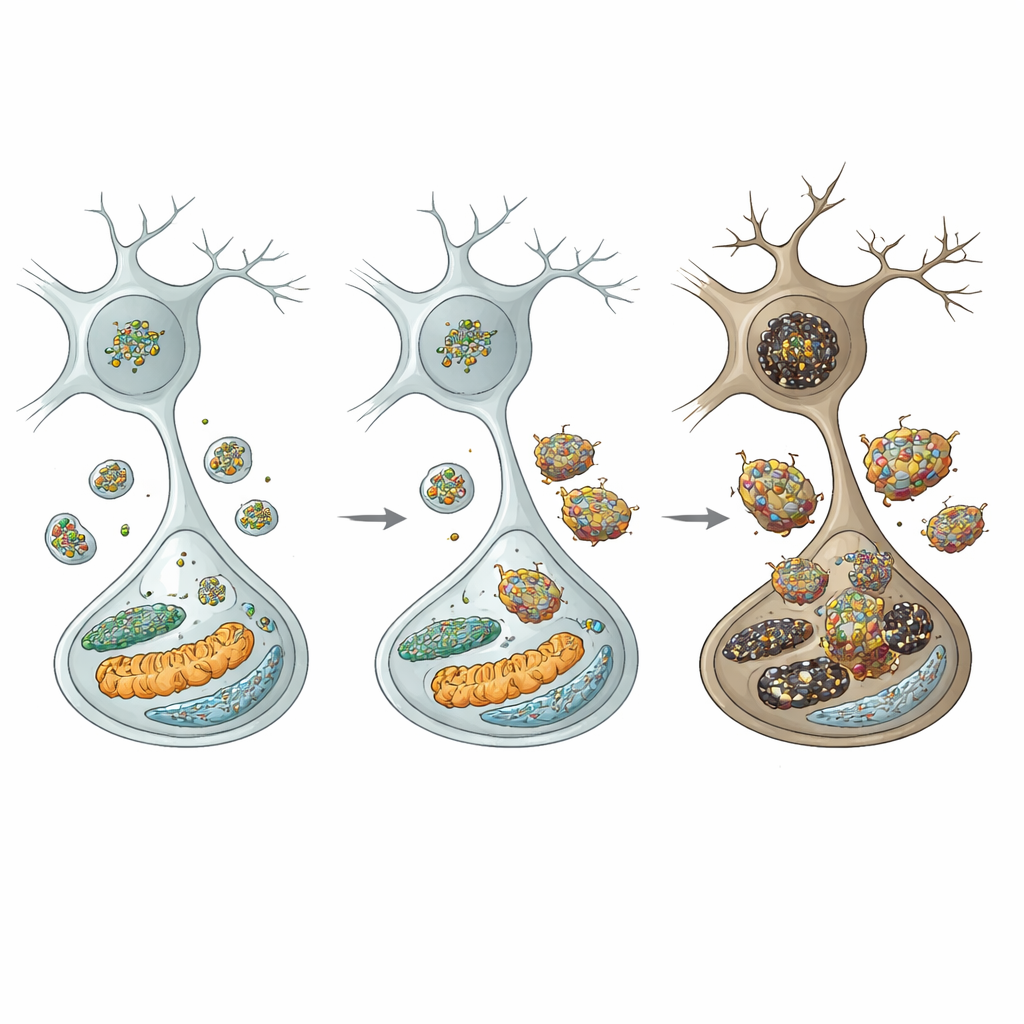
Spreading damage inside and outside the neuron
The excess alpha‑synuclein did not stay put. It accumulated on two vital internal structures—the endoplasmic reticulum and mitochondria—where it triggered stress and damage. In the endoplasmic reticulum, it switched on unfolded‑protein stress responses and pro‑death signals. In mitochondria, it reduced energy‑producing complex I activity, increased reactive oxygen species, and promoted release of cytochrome c, which activates suicide enzymes known as caspases. At the same time, oligomeric alpha‑synuclein likely leaked out of neurons and roused nearby microglia, the brain’s immune sentinels. These microglia shifted into a pro‑inflammatory state, switched on an NLRP3 inflammasome complex, and released inflammatory molecules such as IL‑1β, IL‑18, and TNF‑α. Those signals, in turn, activated additional death pathways inside dopamine neurons, including JNK‑driven and necroptotic routes, creating a vicious circle of injury.
A drug that restores the cell’s cleanup crew
The researchers then asked whether boosting lysosome production could interrupt this cascade. They treated young mutant mice with rapamycin, a drug that indirectly frees a master switch called TFEB to enter the nucleus and turn on lysosome‑building genes, while also stimulating autophagy. After four months, rapamycin‑treated mutant mice showed restored levels of lysosomal markers and cathepsin D in the substantia nigra and much lower alpha‑synuclein buildup. Remarkably, these mice were largely protected from the loss of dopamine neurons in both the substantia nigra and the nearby ventral tegmental area, and their motor performance improved towards normal. By acting upstream on the cell’s waste‑handling network, rapamycin effectively blunted the downstream cascade of protein accumulation, organelle stress, inflammation, and cell death.
What this means for understanding and treating Parkinson’s
Put simply, this study shows how a single inherited fault in a neuron‑specific helper protein can set off a chain reaction: fewer lysosomes, weaker cathepsin D activity, rising levels of toxic alpha‑synuclein, damage to key cellular compartments, inflammatory overreaction, and finally the loss of dopamine neurons that underpins Parkinson’s symptoms. Although this particular mutation is rare, the same themes—lysosomal deficiency, poor protein clearance, and alpha‑synuclein overload—also appear in more common, non‑familial Parkinson’s disease. The work therefore strengthens the idea that restoring the brain’s cleanup systems, for example by TFEB‑activating or autophagy‑boosting drugs like rapamycin or safer successors, could be a promising strategy to slow or prevent Parkinson‑related neurodegeneration.
Citation: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Keywords: Parkinson’s disease, lysosomes, alpha-synuclein, dopaminergic neurons, rapamycin