Clear Sky Science · it
La variante troncatrice PARK19 di Dnajc6 causa upregulation di α-sinucleina patologica indotta da carenza lisosomiale e neurodegenerazione dei neuroni dopaminergici della substantia nigra nei topi knockin PARK19
Perché questo studio sui topi è rilevante per la malattia di Parkinson
La malattia di Parkinson priva le persone del movimento uccidendo lentamente un piccolo gruppo di cellule cerebrali che producono il messaggero chimico dopamina. La maggior parte dei casi deriva da una combinazione di età e ambiente, ma una minoranza è causata da difetti in singoli geni che forniscono indizi potenti sul processo patologico. Questo studio esamina uno di questi geni, DNAJC6, usando un topo ingegnerizzato appositamente per rivelare come una piccola alterazione all’interno delle cellule cerebrali possa compromettere i loro sistemi di smaltimento dei rifiuti, sovraccaricarle con una proteina appiccicosa chiamata alfa-sinucleina e, in ultima analisi, portare alla perdita neuronale e a sintomi simili al Parkinson. 
Una proteina ausiliaria difettosa nelle cellule cerebrali
Il gene DNAJC6 codifica una proteina presente quasi esclusivamente nei neuroni, dove aiuta a gestire la clatrina, una molecola simile a un rivestimento che modella e ricicla le membrane cellulari. In diverse famiglie con una forma a esordio precoce della malattia di Parkinson nota come PARK19, entrambe le copie di DNAJC6 presentano variazioni dannose che recidono un’estremità critica della proteina. Per imitare questa condizione, i ricercatori hanno usato l’editing genico CRISPR per creare topi la cui versione del gene (chiamata Dnajc6 nei topi) si interrompe precocemente in una posizione equivalente. Questi topi omozigoti “Q787X” producono solo la forma accorciata della proteina, mentre i topi eterozigoti portano una copia normale e una mutata, in modo simile ai portatori sani nelle famiglie umane.
Dal difetto genetico ai problemi di movimento
Il gruppo ha seguito i topi con l’avanzare dell’età e ha testato il loro movimento. A cinque mesi tutti i gruppi si comportavano normalmente. Tuttavia, a nove mesi i topi con due copie mutate svilupparono i classici problemi motori simili al Parkinson: si muovevano più lentamente, percorrevano distanze più brevi e impiegavano più tempo a scendere da un palo di prova, rispecchiando ipocinesia e bradicinesia osservate nei pazienti. In esame cerebrale si osservò una marcata perdita di neuroni produttori di dopamina nella substantia nigra e un assottigliamento delle loro terminazioni nervose nello striato. Questi neuroni vulnerabili contenevano inoltre aggregati di alfa-sinucleina fosforilata, analoghi alla patologia dei corpi di Lewy vista nel Parkinson umano. I topi eterozigoti, con una sola copia mutata, non mostrarono questi cambiamenti.
Quando il riciclo cellulare si deteriora
Per comprendere la catena degli eventi, gli autori hanno esaminato i centri di riciclo della cellula: i lisosomi. Nei neuroni dopaminergici dei topi mutanti, i livelli della catena pesante della clatrina erano ridotti, il che dovrebbe interferire con un processo chiamato riformazione lisosomiale autolitica (autophagic lysosome reformation)—il modo in cui le cellule riforniscono il loro pool di lisosomi. Infatti, il numero di lisosomi calò e aumentarono i marcatori di un’autofagia compromessa. In modo cruciale, un enzima lisosomiale chiave, la catepsina D, diminuì, mentre alfa-sinucleina e le sue forme più tossiche, fosforilate e oligomeriche, si accumularono diventando persino resistenti alla degradazione. Questo fallimento lisosomiale fu selettivo: i neuroni inibitori vicini in altre regioni non mostrarono gli stessi deficit, evidenziando la particolare vulnerabilità dei neuroni dopaminergici della substantia nigra. 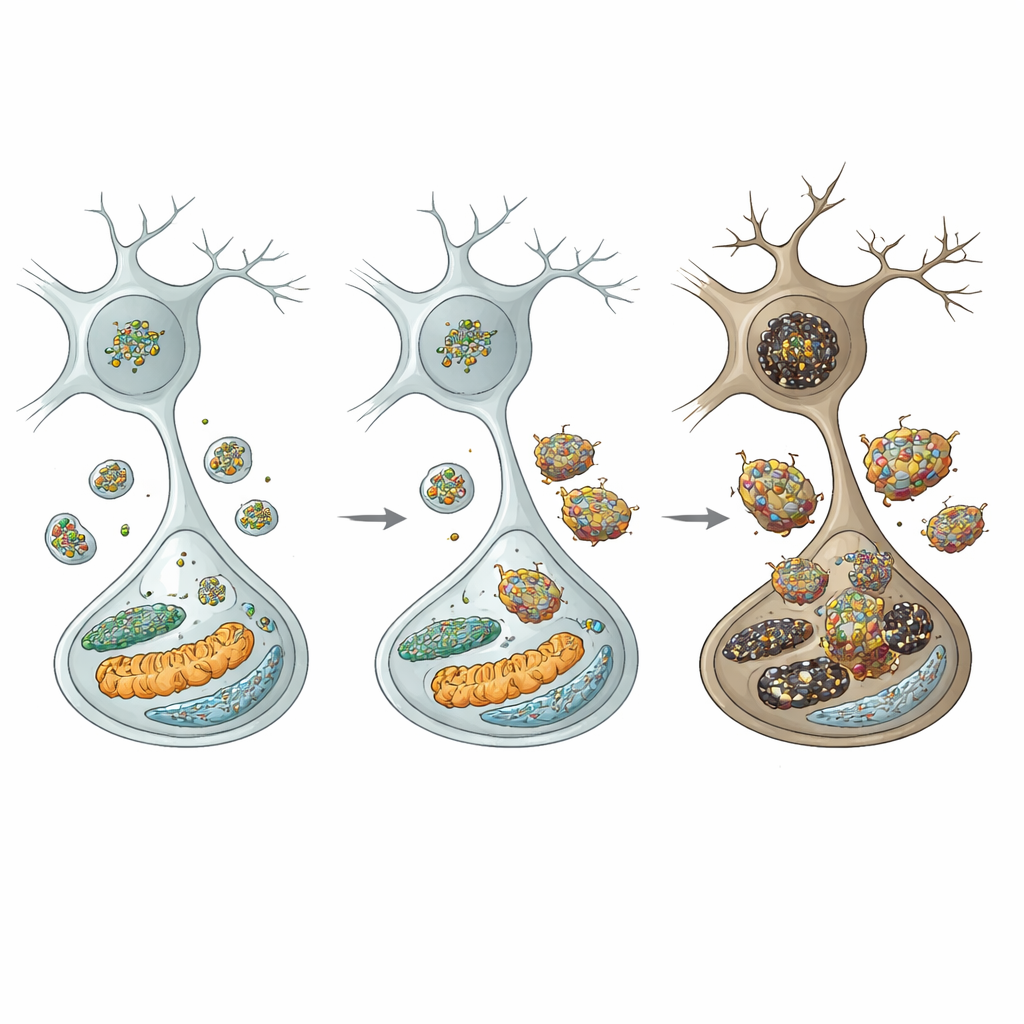
Danno che si propaga dentro e fuori dal neurone
L’eccesso di alfa-sinucleina non rimase confinato. Si accumulò su due strutture interne vitali—il reticolo endoplasmatico e i mitocondri—scatenando stress e danni. Nel reticolo endoplasmatico attivò risposte allo stress da proteine ripiegate e segnali pro‑morte. Nei mitocondri ridusse l’attività del complesso I produttore di energia, aumentò le specie reattive dell’ossigeno e favorì il rilascio di citocromo c, che attiva gli enzimi suicida noti come caspasi. Allo stesso tempo, l’alfa-sinucleina oligomerica probabilmente fuoriuscì dai neuroni e attivò la microglia vicina, le sentinelle immunitarie del cervello. Queste microglia assunsero uno stato pro‑infiammatorio, attivarono il complesso inflammasoma NLRP3 e rilasciarono molecole infiammatorie come IL‑1β, IL‑18 e TNF‑α. Tali segnali, a loro volta, attivarono ulteriori vie di morte all’interno dei neuroni dopaminergici, incluse le rotte guidate da JNK e la necroptosi, creando un circolo vizioso di danno.
Un farmaco che ripristina la squadra di pulizia cellulare
I ricercatori si chiesero quindi se potenziare la produzione di lisosomi potesse interrompere questa cascata. Trattarono giovani topi mutanti con rapamicina, un farmaco che libera indirettamente un interruttore maestro chiamato TFEB permettendogli di entrare nel nucleo e attivare geni per la costruzione dei lisosomi, stimolando al contempo l’autofagia. Dopo quattro mesi, i topi mutanti trattati con rapamicina mostrarono livelli ripristinati di marcatori lisosomiali e di catepsina D nella substantia nigra e un accumulo di alfa-sinucleina molto ridotto. Sorprendentemente, questi topi furono in gran parte protetti dalla perdita di neuroni dopaminergici sia nella substantia nigra sia nell’area tegmentale ventrale adiacente, e le loro prestazioni motorie migliorarono verso la normalità. Agendo a monte sulla rete di gestione dei rifiuti cellulare, la rapamicina attenuò efficacemente la cascata a valle di accumulo proteico, stress degli organelli, infiammazione e morte cellulare.
Cosa significa per comprendere e trattare il Parkinson
In termini semplici, questo studio mostra come un singolo difetto ereditario in una proteina ausiliaria specifica dei neuroni possa innescare una reazione a catena: meno lisosomi, ridotta attività della catepsina D, aumento di alfa-sinucleina tossica, danni a compartimenti cellulari chiave, iperreazione infiammatoria e infine perdita dei neuroni dopaminergici che sottendono i sintomi del Parkinson. Sebbene questa particolare mutazione sia rara, gli stessi temi—carenza lisosomiale, scarsa eliminazione delle proteine e sovraccarico di alfa-sinucleina—appaiono anche nella forma più comune e non familiare del Parkinson. Il lavoro rafforza quindi l’idea che ripristinare i sistemi di pulizia del cervello, per esempio mediante farmaci che attivano TFEB o potenziano l’autofagia come la rapamicina o successori più sicuri, potrebbe essere una strategia promettente per rallentare o prevenire la neurodegenerazione associata al Parkinson.
Citazione: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Parole chiave: Malattia di Parkinson, lisosomi, alfa-sinucleina, neuroni dopaminergici, rapamicina