Clear Sky Science · fr
Le mutant tronqué PARK19 Dnajc6 provoque une déficience lysosomale entraînant une augmentation pathologique de l’α-synucléine et la neurodégénérescence des cellules dopaminergiques de la substance noire chez des souris knockin PARK19
Pourquoi cette étude chez la souris compte pour la maladie de Parkinson
La maladie de Parkinson prive les personnes de leurs mouvements en tuant lentement un petit groupe de cellules cérébrales qui fabriquent le messager chimique dopamine. La majorité des cas résulte d’un mélange d’âge et d’exposition environnementale, mais une minorité est due à des défauts d’un seul gène, qui constituent des indices puissants sur le processus pathologique. Cette étude examine l’un de ces gènes, DNAJC6, en utilisant une souris spécialement conçue pour révéler comment une petite modification à l’intérieur des cellules cérébrales peut paralyser leur système d’élimination des déchets, les surcharger d’une protéine collante appelée alpha‑synucléine et conduire finalement à la perte neuronale et à des symptômes de type Parkinson. 
Une protéine d’assistance défectueuse dans les cellules cérébrales
Le gène DNAJC6 code pour une protéine présente presque exclusivement dans les neurones, où elle aide à gérer la clathrine, une molécule en forme de manteau qui façonne et recycle les membranes cellulaires. Dans plusieurs familles atteintes d’une forme précoce de la maladie de Parkinson connue sous le nom de PARK19, les deux copies de DNAJC6 portent des altérations délétères qui tronquent une extrémité critique de la protéine. Pour reproduire cette situation, les chercheurs ont utilisé l’édition génomique CRISPR pour créer des souris dont la version du gène (appelée Dnajc6 chez la souris) s’arrête précocement en position équivalente. Ces souris homozygotes « Q787X » ne produisent que la forme raccourcie de la protéine, tandis que les souris hétérozygotes portent une copie normale et une copie mutante, semblable aux porteurs sains dans les familles humaines.
Du défaut génétique aux problèmes de mouvement
L’équipe a suivi les souris au fil du vieillissement et a testé leur motricité. À cinq mois, tous les groupes se comportaient normalement. À neuf mois cependant, les souris portant deux copies mutantes ont développé des troubles moteurs classiques ressemblant à la maladie de Parkinson : elles se déplaçaient plus lentement, parcouraient des distances plus courtes et mettaient plus de temps à descendre une tige d’essai, reflétant l’hypokinésie et la bradykinésie observées chez les patients. À l’examen du cerveau, les chercheurs ont constaté une perte marquée des neurones producteurs de dopamine dans la substance noire et un amincissement de leurs terminaisons nerveuses dans le striatum. Ces neurones vulnérables contenaient également des amas d’alpha‑synucléine phosphorylée, similaires à la pathologie des corps de Lewy observée dans la maladie de Parkinson humaine. Les souris hétérozygotes, avec une seule copie mutante, n’ont montré aucun de ces changements.
Quand le recyclage cellulaire se détériore
Pour comprendre la chaîne d’événements, les auteurs ont examiné les centres de recyclage cellulaire : les lysosomes. Dans les neurones dopaminergiques des souris mutantes, les niveaux de la chaîne lourde de la clathrine étaient réduits, ce qui perturbe attendue le processus appelé reformation lysosomale autophagique — la manière dont les cellules reconstituent leur pool de lysosomes. En effet, le nombre de lysosomes a diminué et les marqueurs d’un autophagie déficiente ont augmenté. De façon cruciale, une enzyme lysosomale clé, la cathepsine D, a chuté, tandis que l’alpha‑synucléine et ses formes plus toxiques phosphorylées et oligomériques se sont accumulées et sont même devenues résistantes à la digestion. Cette défaillance lysosomale était sélective : les neurones inhibiteurs voisins dans d’autres régions n’ont pas montré les mêmes déficits, soulignant la vulnérabilité particulière des cellules dopaminergiques de la substance noire. 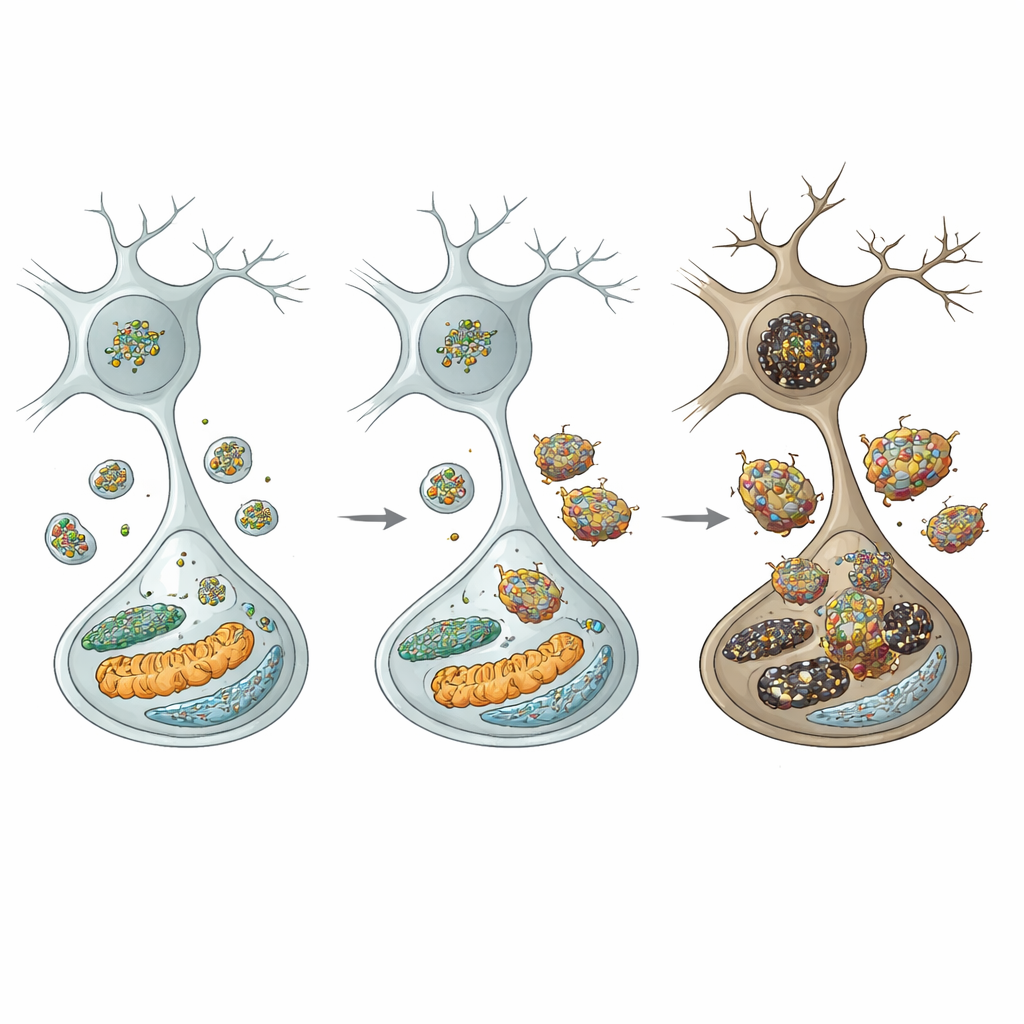
Dommages qui se propagent à l’intérieur et à l’extérieur du neurone
L’excès d’alpha‑synucléine ne restait pas confiné. Il s’est accumulé sur deux structures internes vitales — le réticulum endoplasmique et les mitochondries — où il a déclenché stress et dommages. Dans le réticulum endoplasmique, il a activé les réponses au stress des protéines mal repliées et des signaux pro‑apoptotiques. Dans les mitochondries, il a réduit l’activité du complexe I producteur d’énergie, augmenté les espèces réactives de l’oxygène et favorisé le relargage du cytochrome c, qui active les enzymes suicidaires appelées caspases. Parallèlement, l’alpha‑synucléine oligomérique a probablement fuité hors des neurones et stimulé les microglies voisines, les sentinelles immunitaires du cerveau. Ces microglies sont passées à un état pro‑inflammatoire, ont activé un complexe inflammasome NLRP3 et ont libéré des molécules inflammatoires telles que IL‑1β, IL‑18 et TNF‑α. Ces signaux ont à leur tour activé des voies de mort supplémentaires à l’intérieur des neurones dopaminergiques, y compris des voies dépendantes de JNK et la nécroptose, créant un cercle vicieux de lésions.
Un médicament qui restaure l’équipe de nettoyage cellulaire
Les chercheurs ont ensuite cherché à savoir si stimuler la production de lysosomes pouvait interrompre cette cascade. Ils ont traité de jeunes souris mutantes avec la rapamycine, un médicament qui, de façon indirecte, libère un commutateur maître appelé TFEB pour entrer dans le noyau et activer les gènes de construction des lysosomes, tout en stimulant également l’autophagie. Après quatre mois, les souris mutantes traitées par rapamycine montraient des niveaux restaurés de marqueurs lysosomaux et de cathepsine D dans la substance noire et une accumulation beaucoup plus faible d’alpha‑synucléine. Remarquablement, ces souris étaient en grande partie protégées de la perte de neurones dopaminergiques à la fois dans la substance noire et dans l’aire tegmentale ventrale voisine, et leurs performances motrices s’étaient améliorées vers la normale. En agissant en amont sur le réseau de gestion des déchets cellulaires, la rapamycine a efficacement atténué la cascade en aval d’accumulation protéique, de stress des organites, d’inflammation et de mort cellulaire.
Ce que cela signifie pour la compréhension et le traitement de la maladie de Parkinson
En bref, cette étude montre comment un défaut héréditaire unique dans une protéine d’aide spécifique aux neurones peut déclencher une réaction en chaîne : moins de lysosomes, une activité réduite de la cathepsine D, une augmentation des niveaux d’alpha‑synucléine toxique, des dommages aux compartiments cellulaires clés, une réaction inflammatoire excessive et enfin la perte des neurones dopaminergiques qui sous-tend les symptômes de Parkinson. Bien que cette mutation particulière soit rare, les mêmes thèmes — déficience lysosomale, mauvaise élimination des protéines et surcharge en alpha‑synucléine — apparaissent aussi dans la maladie de Parkinson plus fréquente et non familiale. Ces travaux renforcent donc l’idée que restaurer les systèmes de nettoyage du cerveau, par exemple via des médicaments activant TFEB ou stimulant l’autophagie comme la rapamycine ou des dérivés plus sûrs, pourrait être une stratégie prometteuse pour ralentir ou prévenir la neurodégénérescence liée à Parkinson.
Citation: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Mots-clés: Maladie de Parkinson, lysosomes, alpha-synucléine, neurones dopaminergiques, rapamycine