Clear Sky Science · pt
Mutante truncado PARK19 Dnajc6 causa aumento de α-sinucleína patológica induzido por deficiência lisossomal e neurodegeneração de células dopaminérgicas da substância negra em camundongos knockin PARK19
Por que este estudo em camundongos importa para a doença de Parkinson
A doença de Parkinson priva as pessoas de movimento ao matar lentamente um pequeno grupo de células cerebrais que produzem o mensageiro químico dopamina. A maioria dos casos resulta de uma combinação de idade e ambiente, mas uma minoria é causada por falhas em um único gene que funcionam como pistas poderosas sobre o processo da doença. Este estudo examina um desses genes, DNAJC6, usando um camundongo especialmente modificado para revelar como uma pequena alteração no interior das células cerebrais pode prejudicar seu sistema de descarte de resíduos, sobrecarregá‑las com uma proteína pegajosa chamada alfa‑sinucleína e, em última instância, levar à perda neuronal e a sintomas semelhantes aos da doença de Parkinson. 
Uma proteína auxiliar defeituosa nas células cerebrais
O gene DNAJC6 produz uma proteína encontrada quase exclusivamente em neurônios, onde ajuda a gerir a clatrina, uma molécula em forma de “casco” que molda e recicla membranas celulares. Em várias famílias com uma forma de início precoce da doença de Parkinson conhecida como PARK19, ambas as cópias de DNAJC6 carregam alterações deletérias que cortam uma extremidade crítica da proteína. Para imitar essa situação, os pesquisadores usaram edição genética CRISPR para criar camundongos cuja versão do gene (chamada Dnajc6 em camundongos) termina precocemente em uma posição equivalente. Esses camundongos homozigotos “Q787X” produzem apenas a forma encurtada da proteína, enquanto camundongos heterozigotos carregam uma cópia normal e uma mutante, semelhante a portadores saudáveis em famílias humanas.
Do defeito genético aos problemas de movimento
A equipe acompanhou os camundongos com o envelhecimento e testou seus movimentos. Aos cinco meses, todos os grupos comportaram‑se normalmente. Aos nove meses, no entanto, os camundongos com duas cópias mutantes desenvolveram problemas motores clássicos semelhantes aos da doença de Parkinson: moviam‑se mais lentamente, percorreram distâncias menores e levaram mais tempo para descer um poste de teste, espelhando a hipocinesia e a bradicinesia observadas em pacientes. Quando os cientistas examinaram seus cérebros, observaram uma perda marcante de neurônios produtores de dopamina na substância negra e afinamento de seus terminais nervosos no estriado. Esses neurônios vulneráveis também continham aglomerados de alfa‑sinucleína fosforilada, semelhantes à patologia de corpos de Lewy vista na doença de Parkinson humana. Camundongos heterozigotos, com apenas uma cópia mutante, não mostraram essas alterações.
Quando a reciclagem celular entra em colapso
Para entender a cadeia de eventos, os autores analisaram os centros de reciclagem da célula: os lisossomos. Em neurônios dopaminérgicos de camundongos mutantes, os níveis da cadeia pesada da clatrina estavam reduzidos, o que deve perturbar um processo chamado reforma lisossomal por autofagia—o modo como as células reabastecem seu conjunto de lisossomos. De fato, o número de lisossomos diminuiu, e marcadores de autofagia comprometida aumentaram. Crucialmente, uma enzima lisossomal chave, a catepsina D, caiu, enquanto alfa‑sinucleína e suas formas mais tóxicas fosforiladas e oligoméricas se acumularam e até se tornaram resistentes à digestão. Essa falha lisossomal foi seletiva: neurônios inibitórios vizinhos em outras regiões não mostraram os mesmos déficits, destacando a vulnerabilidade particular das células dopaminérgicas da substância negra. 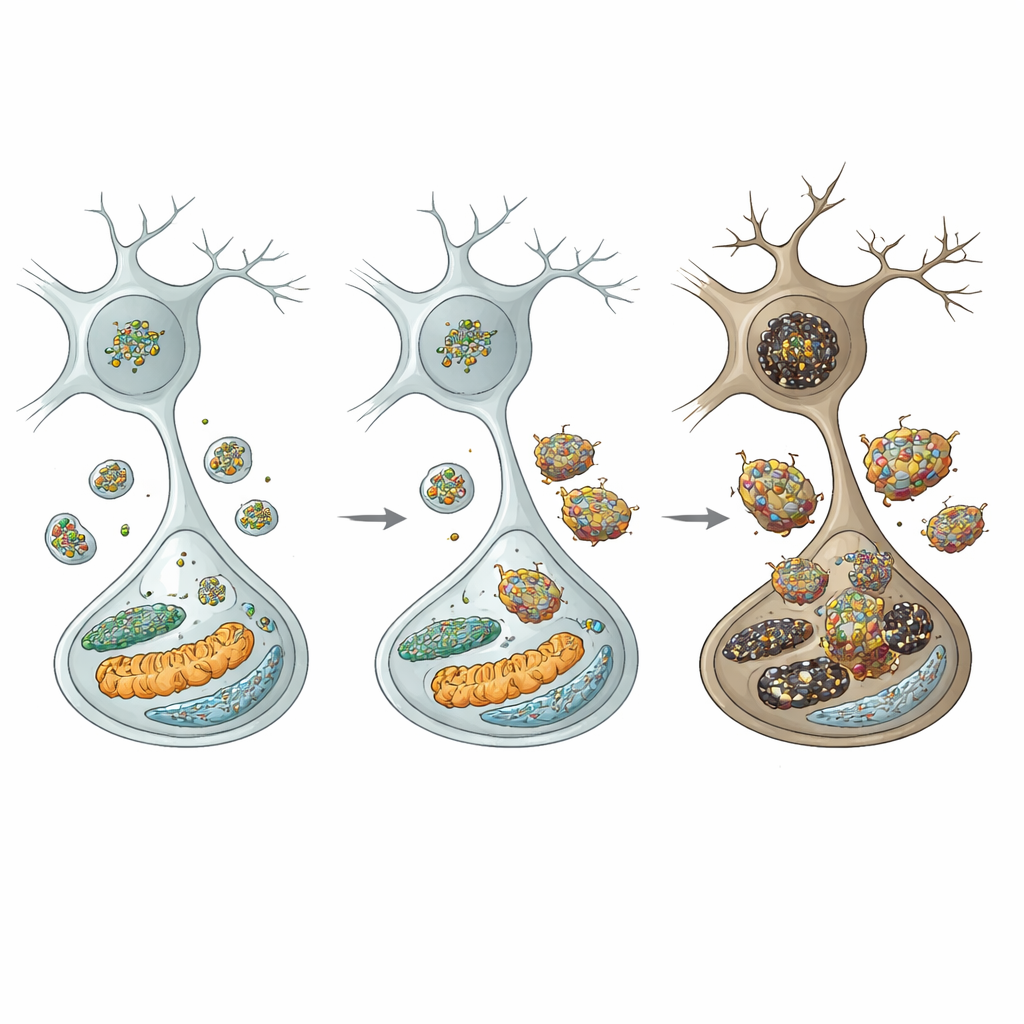
Dano que se espalha dentro e fora do neurônio
A alfa‑sinucleína em excesso não ficou localizada. Ela se acumulou em duas estruturas internas vitais—o retículo endoplasmático e as mitocôndrias—onde desencadeou estresse e dano. No retículo endoplasmático, ativou respostas ao acúmulo de proteínas não dobradas e sinais pró‑morte. Nas mitocôndrias, reduziu a atividade do complexo I produtor de energia, aumentou espécies reativas de oxigênio e promoveu a liberação de citocromo c, que ativa enzimas suicidas conhecidas como caspases. Ao mesmo tempo, a alfa‑sinucleína oligomérica provavelmente vazou para fora dos neurônios e ativou micróglias próximas, os sentinelas imunes do cérebro. Essas micróglias migraram para um estado pró‑inflamatório, ativaram o complexo inflamassoma NLRP3 e liberaram moléculas inflamatórias como IL‑1β, IL‑18 e TNF‑α. Esses sinais, por sua vez, ativaram vias adicionais de morte dentro dos neurônios dopaminérgicos, incluindo rotas conduzidas por JNK e necroptose, criando um círculo vicioso de lesão.
Um fármaco que restaura a equipe de limpeza celular
Os pesquisadores então perguntaram se impulsionar a produção de lisossomos poderia interromper essa cascata. Trataram camundongos mutantes jovens com rapamicina, um fármaco que libera indiretamente um interruptor mestre chamado TFEB para entrar no núcleo e ativar genes de construção de lisossomos, enquanto também estimula a autofagia. Após quatro meses, camundongos mutantes tratados com rapamicina mostraram níveis restaurados de marcadores lisossomais e de catepsina D na substância negra e acúmulo muito menor de alfa‑sinucleína. Notavelmente, esses camundongos ficaram amplamente protegidos da perda de neurônios dopaminérgicos tanto na substância negra quanto na área tegmental ventral próxima, e seu desempenho motor melhorou em direção ao normal. Ao agir a montante na rede de manejo de resíduos da célula, a rapamicina atenuou efetivamente a cascata subsequente de acúmulo proteico, estresse em organelas, inflamação e morte celular.
O que isso significa para entender e tratar o Parkinson
Em poucas palavras, este estudo mostra como uma única falha herdada em uma proteína auxiliar específica de neurônio pode desencadear uma reação em cadeia: menos lisossomos, atividade reduzida de catepsina D, níveis crescentes de alfa‑sinucleína tóxica, dano a compartimentos celulares-chave, reação inflamatória exagerada e, finalmente, a perda de neurônios dopaminérgicos que sustenta os sintomas do Parkinson. Embora essa mutação particular seja rara, os mesmos temas—deficiência lisossomal, clareza proteica deficiente e sobrecarga de alfa‑sinucleína—também aparecem na doença de Parkinson mais comum e não familiar. O trabalho, portanto, fortalece a ideia de que restaurar os sistemas de limpeza do cérebro, por exemplo com fármacos que ativem TFEB ou aumentem a autofagia como a rapamicina ou sucessores mais seguros, pode ser uma estratégia promissora para retardar ou prevenir a neurodegeneração relacionada ao Parkinson.
Citação: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Palavras-chave: Doença de Parkinson, lisossomos, alfa-sinucleína, neurônios dopaminérgicos, rapamicina