Clear Sky Science · de
Trunkierungs-Mutante PARK19 Dnajc6 verursacht lysosomale Defizienz‑induzierte Hochregulierung pathologischen α‑Synucleins und Neurodegeneration dopaminerger Zellen der Substantia nigra in PARK19‑Knockin‑Mäusen
Warum diese Mausstudie für die Parkinson‑Krankheit wichtig ist
Die Parkinson‑Krankheit beraubt Menschen ihrer Bewegungsfähigkeit, weil sie langsam eine kleine Gruppe von Gehirnzellen zerstört, die den Botenstoff Dopamin produzieren. Die meisten Fälle entstehen durch eine Mischung aus Alter und Umwelt, doch eine Minderheit wird durch einzelne Genfehler verursacht, die starke Hinweise auf die Krankheitsmechanismen liefern. Diese Studie untersucht eines dieser Gene, DNAJC6, mithilfe eines speziell gezüchteten Mausmodells, um zu zeigen, wie eine winzige Veränderung tief in den Zellen die Abfallentsorgung aushebeln, die Zellen mit einem klebrigen Protein namens Alpha‑Synuclein überfrachten und letztlich zum Neuronenverlust und zu Parkinson‑ähnlichen Symptomen führen kann. 
Ein fehlerhaftes Helferprotein in Nervenzellen
Das Gen DNAJC6 produziert ein Protein, das fast ausschließlich in Neuronen vorkommt und dort an der Steuerung von Clathrin mitwirkt, einem mantelartigen Molekül, das Zellmembranen formt und recycelt. In mehreren Familien mit einer früh einsetzenden Form der Parkinson‑Krankheit, bekannt als PARK19, tragen beide Kopien von DNAJC6 schädigende Veränderungen, die ein kritisches Ende des Proteins abschneiden. Um diese Situation nachzubilden, erzeugten die Forscher mithilfe von CRISPR‑Geneditierung Mäuse, deren Genversion (bei Mäusen Dnajc6 genannt) an einer äquivalenten Stelle vorzeitig abbricht. Diese homozygoten „Q787X“‑Mäuse produzieren nur die verkürzte Form des Proteins, während heterozygote Mäuse eine normale und eine mutierte Kopie tragen, ähnlich wie gesunde Träger in betroffenen menschlichen Familien.
Vom Gendefekt zu Bewegungsstörungen
Das Team verfolgte die Mäuse im Alter und testete ihre Motorik. Mit fünf Monaten verhielten sich alle Gruppen normal. Mit neun Monaten entwickelten Mäuse mit zwei mutierten Kopien jedoch klassische Parkinson‑artige Bewegungsstörungen: Sie bewegten sich langsamer, legten kürzere Strecken zurück und benötigten länger, um eine Teststange hinunterzuklettern, was Hypokinese und Bradykinese beim Patienten widerspiegelt. Bei der Untersuchung der Gehirne stellten die Wissenschaftler einen deutlichen Verlust dopaminproduzierender Neurone in der Substantia nigra sowie eine Ausdünnung ihrer Nervenendigungen im Striatum fest. Diese verletzlichen Neurone enthielten zudem Aggregate phosphorylierten Alpha‑Synucleins, vergleichbar mit den Lewy‑Body‑Pathologien bei der menschlichen Parkinson‑Krankheit. Heterozygote Mäuse mit nur einer mutierten Kopie zeigten keine dieser Veränderungen.
Wenn das zelluläre Recycling versagt
Um die Ereigniskette zu verstehen, betrachteten die Autorinnen und Autoren die Recyclingzentren der Zelle: die Lysosomen. In Dopaminneuronen der mutanten Mäuse waren die Pegel der schweren Clathrin‑Untereinheit reduziert, was erwartungsgemäß einen Prozess namens autophagische Lysosomenreformation stören würde — den Mechanismus, mit dem Zellen ihren Lysosomenbestand erneuern. Tatsächlich sank die Zahl der Lysosomen, und Marker für beeinträchtigte zelluläre „Selbstverzehrung“ (Autophagie) stiegen an. Entscheidend nahm ein wichtiger lysosomaler Enzymbestandteil, Cathepsin D, ab, während Alpha‑Synuclein und seine toxischeren phosphorylierten und oligomeren Formen akkumulierten und sogar resistent gegen Abbau wurden. Dieses lysosomale Versagen war selektiv: benachbarte inhibitorische Neurone in anderen Regionen zeigten nicht dieselben Defizite, was die besondere Verwundbarkeit der Dopaminzellen der Substantia nigra hervorhebt. 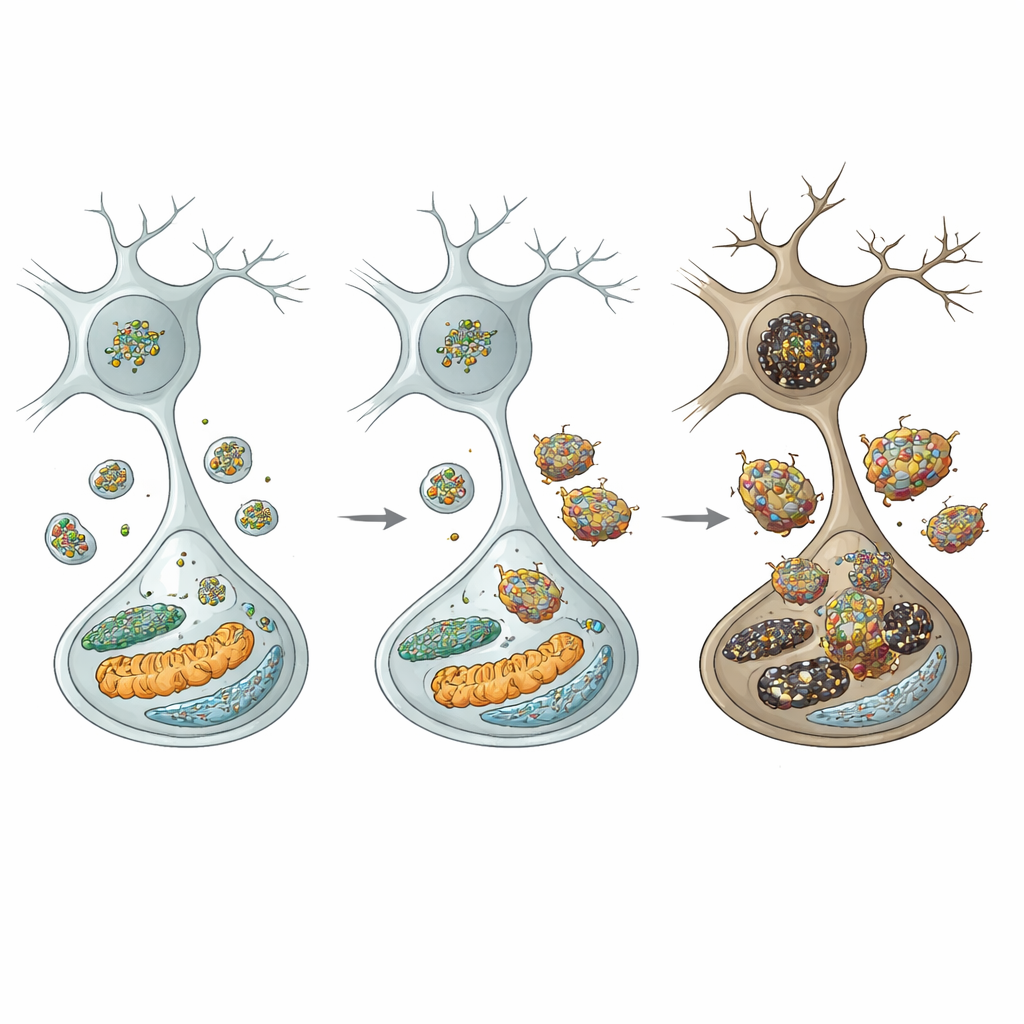
Ausbreitende Schäden innerhalb und außerhalb der Zelle
Das überschüssige Alpha‑Synuclein blieb nicht lokal begrenzt. Es reicherte sich auf zwei wichtigen inneren Strukturen an — dem endoplasmatischen Retikulum und den Mitochondrien — und löste dort Stress und Schäden aus. Im endoplasmatischen Retikulum aktivierte es die Reaktionen auf fehlgefaltete Proteine und pro‑apoptotische Signale. In den Mitochondrien verringerte es die Aktivität des Energie erzeugenden Komplexes I, erhöhte reaktive Sauerstoffspezies und förderte die Freisetzung von Cytochrom c, das Suizid‑Enzyme namens Caspasen aktiviert. Gleichzeitig traten oligomere Alpha‑Synuclein‑Formen wahrscheinlich aus den Neuronen aus und aktivierten benachbarte Mikroglia, die immunologischen Wächter des Gehirns. Diese Mikroglia gingen in einen pro‑inflammatorischen Zustand über, schalteten das NLRP3‑Inflammasom ein und setzten entzündungsfördernde Moleküle wie IL‑1β, IL‑18 und TNF‑α frei. Diese Signale aktivierten wiederum zusätzliche Todeswege innerhalb der Dopaminneurone, darunter JNK‑getriebene und nekroptotische Pfade, wodurch ein teuflischer Kreis der Schädigung entstand.
Ein Wirkstoff, der das zelluläre Aufräumteam wiederherstellt
Die Forscher fragten dann, ob die Anhebung der Lysosomenproduktion diese Kaskade unterbrechen könne. Sie behandelten junge mutante Mäuse mit Rapamycin, einem Wirkstoff, der indirekt einen Masterregulator namens TFEB freisetzt, damit dieser in den Zellkern gelangt und lysosomenaufbauende Gene aktiviert, während gleichzeitig die Autophagie angeregt wird. Nach vier Monaten zeigten rapamycin‑behandelte mutante Mäuse wiederhergestellte Pegel lysosomaler Marker und von Cathepsin D in der Substantia nigra sowie deutlich geringere Alpha‑Synuclein‑Akkumulation. Bemerkenswerterweise waren diese Mäuse weitgehend vor dem Verlust von Dopaminneuronen sowohl in der Substantia nigra als auch im benachbarten ventralen tegmentalen Gebiet geschützt, und ihre motorische Leistung näherte sich dem Normalzustand an. Indem Rapamycin stromaufwärts in das zelluläre Abfallentsorgungssystem eingriff, dämpfte es effektiv die nachgelagerten Prozesse von Proteinakkumulation, Organelstress, Entzündung und Zelltod.
Was das für das Verständnis und die Behandlung von Parkinson bedeutet
Kurz gesagt zeigt diese Studie, wie ein einziger vererbter Fehler in einem neuronenspezifischen Helferprotein eine Kettenreaktion auslösen kann: weniger Lysosomen, schwächere Cathepsin‑D‑Aktivität, steigende Mengen toxischen Alpha‑Synucleins, Schädigung wichtiger Zellkompartimente, übermäßige Entzündungsreaktionen und schließlich der Verlust von Dopaminneuronen, der den Kern der Parkinson‑Symptomatik bildet. Obwohl diese spezielle Mutation selten ist, treten dieselben Themen — lysosomale Defizienz, mangelnde Proteinentfernung und Alpha‑Synuclein‑Überladung — auch bei der häufigeren, nicht‑familiären Parkinson‑Krankheit auf. Die Arbeit stärkt daher die Idee, dass die Wiederherstellung der Aufräumsysteme des Gehirns, etwa durch TFEB‑aktivierende oder Autophagie‑stärkende Wirkstoffe wie Rapamycin oder sicherere Nachfolger, eine vielversprechende Strategie sein könnte, um Parkinson‑bedingte Neurodegeneration zu verlangsamen oder zu verhindern.
Zitation: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Schlüsselwörter: Parkinson-Krankheit, Lysosomen, Alpha‑Synuclein, Dopaminerge Neurone, Rapamycin