Clear Sky Science · ru
Усечённый мутант PARK19 Dnajc6 вызывает дефицит лизосом, приводящий к повышению патологического α-синуклеина и нейродегенерации дофаминергических клеток черной субстанции в кинотах PARK19
Почему это исследование на мышах важно для болезни Паркинсона
Болезнь Паркинсона лишает людей движения, постепенно убивая небольшую группу клеток мозга, производящих химический мессенджер дофамин. В большинстве случаев заболевание развивается под влиянием возраста и окружающей среды, но в некоторых оно вызывается единичными генетическими поражениями, которые дают важные подсказки о механизмах болезни. В этом исследовании изучают один из таких генов — DNAJC6 — на специально созданных мышах, чтобы показать, как малое изменение внутри клеток мозга может подорвать их систему утилизации отходов, перегрузить их липким белком α‑синуклеином и в конечном счёте привести к гибели нейронов и симптомам, похожим на Паркинсон.
Сбой помощника белка в нейронах
Ген DNAJC6 кодирует белок, обнаруживающийся почти исключительно в нейронах, где он участвует в регулировании клатрина — «оболочечного» молекулы, формирующей и перерабатывающей клеточные мембраны. У нескольких семей с ранним началом болезни Паркинсона, известной как PARK19, обе копии DNAJC6 несут повреждения, отрезающие критический участок конца белка. Чтобы воспроизвести эту ситуацию, исследователи с помощью редактирования генома CRISPR создали мышей, у которых версия гена (в мышах называемая Dnajc6) заканчивается преждевременно в эквивалентном положении. Эти гомозиготные «Q787X» мыши синтезируют только усечённую форму белка, тогда как гетерозиготные животные несут одну нормальную и одну мутантную копию, подобно здоровым носителям в человеческих семьях.
От генетической ошибки к проблемам с движением
Команда наблюдала за мышами по мере их старения и тестировала двигательные функции. В пяти месяцев все группы вели себя нормально. Однако к девяти месяцам мыши с двумя мутантными копиями развили классические моторные нарушения, похожие на Паркинсона: они двигались медленнее, покрывали меньшие расстояния и требовали больше времени, чтобы сойти по тестовой палке, отражая гипокинезию и брадикенезию у пациентов. При исследовании мозга ученые обнаружили заметную потерю дофаминпродуцирующих нейронов в substantia nigra и истончение их нервных окончаний в стриатуме. В уязвимых нейронах также обнаруживались скопления фосфорилированного α‑синуклеина, подобные патологии телец Леви при человеческой болезни Паркинсона. Гетерозиготные мыши с одной мутантной копией таких изменений не демонстрировали.
Когда клеточный «мусоропереработчик» выходит из строя
Чтобы проследить последовательность событий, авторы изучили «перерабатывающие» центры клетки — лизосомы. В дофаминергических нейронах мутантных мышей уровень тяжёлой цепи клатрина был снижён, что, как ожидалось, нарушает процесс, называемый аутолизосомным восстановлением (autophagic lysosome reformation) — способ, которым клетки пополняют пул лизосом. Действительно, число лизосом уменьшилось, и возросли маркёры нарушенного клеточного «самопоедания» (аутофагии). Критически важный лизосомный фермент катепсин D уменьшился, тогда как α‑синуклеин и его более токсичные фосфорилированные и олигомерные формы накапливались и даже стали устойчивы к перевариванию. Этот лизосомный сбой был селективным: соседние ингибирующие нейроны в других областях не проявляли таких дефицитов, подчёркивая особую уязвимость дофаминовых клеток substantia nigra.
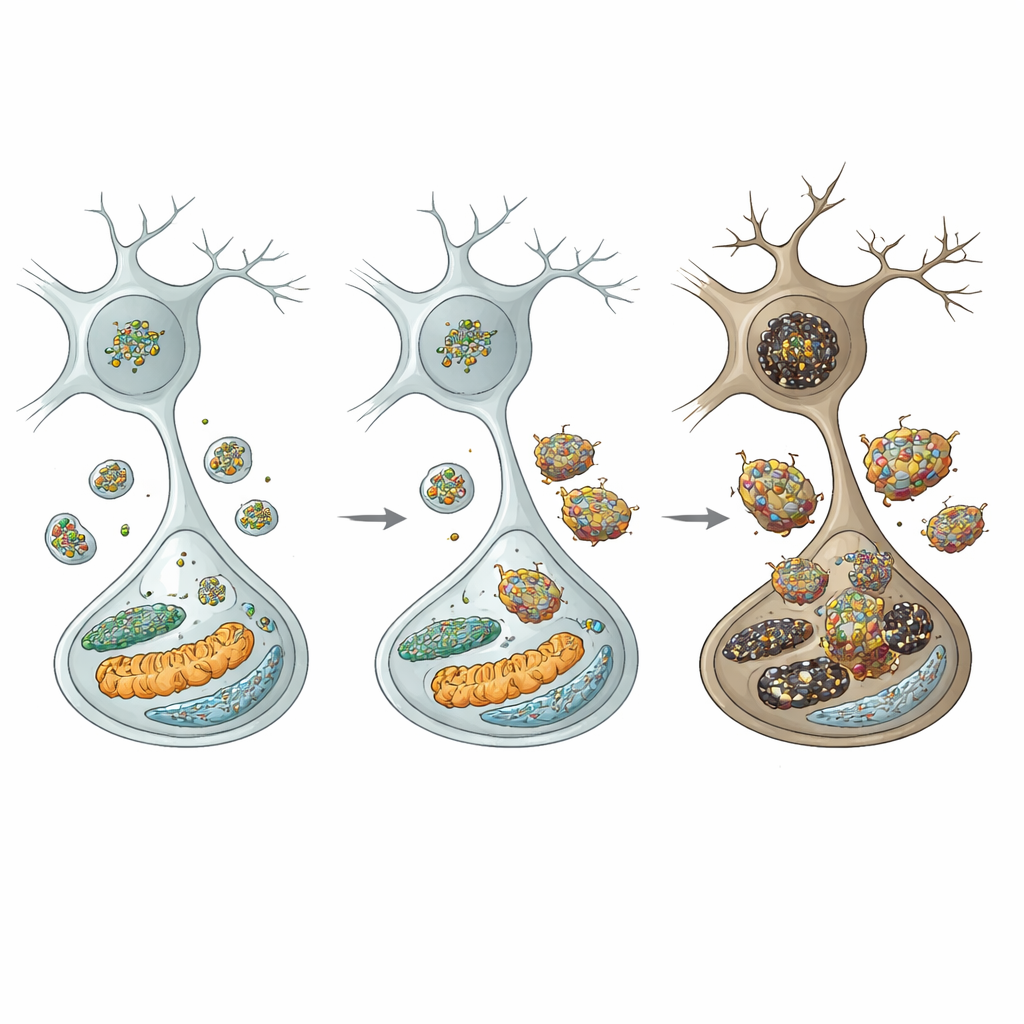
Распространение повреждений внутри и вне нейрона
Избыточный α‑синуклеин не оставался на месте. Он накапливался на двух важных внутренних структурах — эндоплазматическом ретикулуме и митохондриях — где вызывал стресс и повреждение. В эндоплазматическом ретикулуме это активировало ответы на неправильно свернутые белки и профатальные сигналы. В митохондриях снижалась активность комплекса I, увеличивалось образование реактивных форм кислорода и стимулировался выход цитохрома c, который запускает супервоспроизводящие ферменты — каспазы. Одновременно олигомерный α‑синуклеин, вероятно, просачивался из нейронов и активировал соседние микроглии — иммунные стражи мозга. Эти микроглии переключались в провоспалительное состояние, активировали инфламмасому NLRP3 и выделяли воспалительные молекулы, такие как IL‑1β, IL‑18 и TNF‑α. Эти сигналы, в свою очередь, запускали дополнительные пути гибели внутри дофаминергических нейронов, включая JNK‑опосредованные и некроптозные механизмы, создавая порочный круг повреждения.
Препарат, восстанавливающий «уборочную бригаду» клетки
Далее исследователи проверили, можно ли прервать эту каскаду, усилив образование лизосом. Молодых мутантных мышей лечили рапамицином — препаратом, который косвенно освобождает мастер‑регулятор TFEB, позволяя ему войти в ядро и включить гены построения лизосом, а также стимулировать аутофагию. Через четыре месяца у мутантных мышей, получавших рапамицин, восстановились уровни лизосомальных маркёров и катепсина D в substantia nigra, а накопление α‑синуклеина значительно уменьшилось. Примечательно, что эти мыши в значительной степени были защищены от потери дофаминовых нейронов как в substantia nigra, так и в соседней вентральной тегментальной области, а их моторика улучшилась по направлению к норме. Воздействуя верхнеуровнево на сеть утилизации клеточных отходов, рапамицин эффективно притупил последующую каскаду накопления белка, стресса органелл, воспаления и гибели клеток.
Что это значит для понимания и лечения болезни Паркинсона
Проще говоря, это исследование демонстрирует, как одна наследственная ошибка в нейрон‑специфическом «помощнике» белка может запустить цепную реакцию: уменьшение числа лизосом, ослабление активности катепсина D, рост уровней токсичного α‑синуклеина, повреждение ключевых клеточных компартментов, воспалительная сверхреакция и, в конце концов, потеря дофаминовых нейронов, лежащая в основе симптомов Паркинсона. Хотя эта конкретная мутация редка, те же темы — дефицит лизосом, плохая очистка белков и перегрузка α‑синуклеином — наблюдаются и при более распространённых, нефамильных формах болезни Паркинсона. Работа усиливает идею о том, что восстановление «уборочных» систем мозга, например с помощью активаторов TFEB или препаратов, усиливающих аутофагию, таких как рапамицин или более безопасные его аналоги, может быть перспективной стратегией замедления или предотвращения паркинсоновской нейродегенерации.
Цитирование: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Ключевые слова: Болезнь Паркинсона, лизосомы, альфа-синуклеин, дофаминергические нейроны, рапамицин