Clear Sky Science · es
La mutación truncante PARK19 en Dnajc6 provoca deficiencia lisosomal que induce la acumulación patológica de α-sinucleína y la neurodegeneración de las células dopaminérgicas de la sustancia negra en ratones knockin PARK19
Por qué este estudio en ratones importa para la enfermedad de Parkinson
La enfermedad de Parkinson priva a las personas del movimiento al matar lentamente a un pequeño grupo de células cerebrales que producen el mensajero químico dopamina. La mayoría de los casos surgen de una mezcla de edad y entorno, pero una minoría se debe a fallos en un solo gen que actúan como pistas poderosas sobre el proceso de la enfermedad. Este estudio examina uno de esos genes, DNAJC6, usando un ratón especialmente diseñado para revelar cómo un pequeño cambio en el interior de las neuronas puede paralizar sus sistemas de eliminación de desechos, sobrecargarlas con una proteína pegajosa llamada alfa-sinucleína y, en última instancia, conducir a la pérdida de neuronas y a síntomas similares a los del Parkinson. 
Una proteína auxiliar defectuosa en las neuronas
El gen DNAJC6 produce una proteína que se encuentra casi exclusivamente en las neuronas, donde ayuda a gestionar la clatrina, una molécula en forma de cubierta que modela y recicla las membranas celulares. En varias familias con una forma de inicio temprano de la enfermedad de Parkinson conocida como PARK19, las dos copias de DNAJC6 presentan cambios perjudiciales que recortan un extremo crítico de la proteína. Para imitar esta situación, los investigadores usaron edición genética CRISPR para crear ratones cuya versión del gen (llamada Dnajc6 en ratones) se detiene antes en una posición equivalente. Estos ratones homocigotos “Q787X” producen solo la forma acortada de la proteína, mientras que los heterocigotos llevan una copia normal y una mutante, similar a los portadores sanos en familias humanas.
Del defecto genético a los problemas de movimiento
El equipo siguió a los ratones mientras envejecían y evaluó su motricidad. A los cinco meses, todos los grupos se comportaban con normalidad. Sin embargo, a los nueve meses, los ratones con dos copias mutantes desarrollaron problemas motores clásicos similares al Parkinson: se movían más despacio, recorrían distancias más cortas y necesitaban más tiempo para descender un poste en las pruebas, lo que refleja hipocinesia y bradicinesia en pacientes. Al examinar sus cerebros, observaron una pérdida notable de neuronas productoras de dopamina en la sustancia negra y un adelgazamiento de sus terminales nerviosas en el estriado. Estas neuronas vulnerables también contenían agregados de alfa-sinucleína fosforilada, análogos a la patología de cuerpos de Lewy observada en la enfermedad de Parkinson humana. Los ratones heterocigotos, con solo una copia mutante, no mostraron ninguno de estos cambios.
Cuando el reciclaje celular se descompone
Para entender la cadena de eventos, los autores analizaron los centros de reciclaje celular: los lisosomas. En las neuronas dopaminérgicas de los ratones mutantes, los niveles de la cadena pesada de clatrina estaban reducidos, lo que se espera que perturbe un proceso llamado reformación lisosomal autófaga—la manera en que las células reponen su reserva de lisosomas. Efectivamente, el número de lisosomas disminuyó y los marcadores de autocatabolismo (autofagia) deficiente aumentaron. De manera crucial, una enzima lisosomal clave, la catepsina D, se redujo, mientras que la alfa-sinucleína y sus formas más tóxicas fosforiladas y oligoméricas se acumularon e incluso se volvieron resistentes a la digestión. Esta falla lisosomal fue selectiva: las neuronas inhibitorias vecinas en otras regiones no mostraron los mismos déficits, lo que subraya la vulnerabilidad particular de las células dopaminérgicas de la sustancia negra. 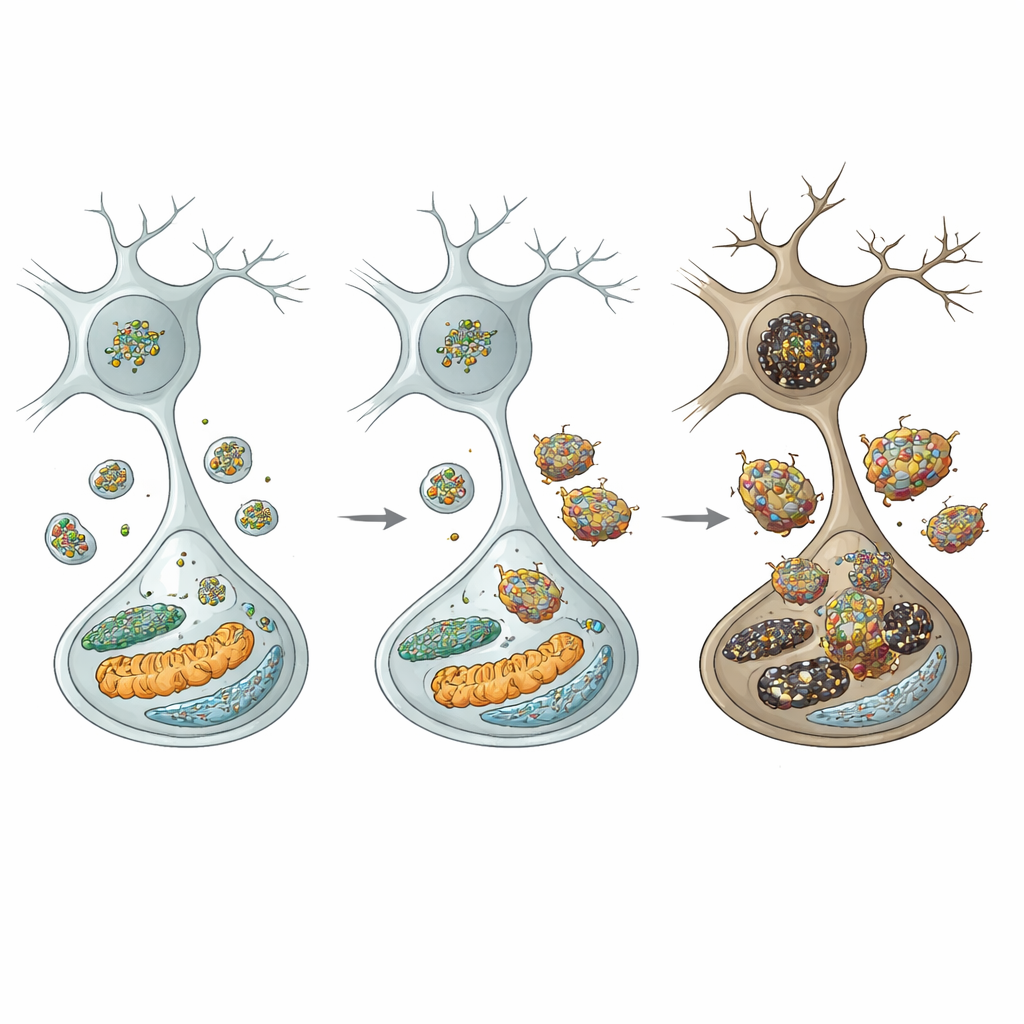
Daño que se propaga dentro y fuera de la neurona
La alfa-sinucleína en exceso no permaneció localizada. Se acumuló en dos estructuras internas vitales—el retículo endoplásmico y las mitocondrias—donde desencadenó estrés y daño. En el retículo endoplásmico activó respuestas de estrés por proteínas mal plegadas y señales pro-muerte. En las mitocondrias redujo la actividad del complejo I productor de energía, aumentó las especies reactivas de oxígeno y promovió la liberación de citocromo c, que activa enzimas suicidas conocidas como caspasas. Al mismo tiempo, la alfa-sinucleína oligomérica probablemente se filtró fuera de las neuronas y activó a las microglías cercanas, los centinelas inmunitarios del cerebro. Estas microglías cambiaron a un estado proinflamatorio, activaron el complejo inflammasoma NLRP3 y liberaron moléculas inflamatorias como IL-1β, IL-18 y TNF-α. Esas señales, a su vez, activaron vías adicionales de muerte dentro de las neuronas dopaminérgicas, incluidas rutas impulsadas por JNK y necroptosis, creando un círculo vicioso de lesión.
Un fármaco que restaura el equipo de limpieza celular
Los investigadores se preguntaron entonces si potenciar la producción de lisosomas podría interrumpir esta cascada. Trataron a ratones mutantes jóvenes con rapamicina, un fármaco que libera indirectamente un interruptor maestro llamado TFEB para que entre en el núcleo y active genes constructores de lisosomas, además de estimular la autofagia. Tras cuatro meses, los ratones mutantes tratados con rapamicina mostraron niveles restaurados de marcadores lisosomales y de catepsina D en la sustancia negra y una acumulación de alfa-sinucleína mucho menor. De forma notable, estos ratones estuvieron en gran parte protegidos de la pérdida de neuronas dopaminérgicas tanto en la sustancia negra como en el área tegmental ventral cercana, y su rendimiento motor mejoró hacia la normalidad. Al actuar aguas arriba sobre la red de manejo de desechos de la célula, la rapamicina mitigó eficazmente la cascada posterior de acumulación proteica, estrés orgánelar, inflamación y muerte celular.
Qué significa esto para entender y tratar el Parkinson
En términos simples, este estudio muestra cómo una sola anomalía heredada en una proteína auxiliar específica de las neuronas puede desencadenar una reacción en cadena: menos lisosomas, menor actividad de catepsina D, aumento de niveles de alfa-sinucleína tóxica, daño a compartimentos celulares clave, sobrerreacción inflamatoria y, finalmente, pérdida de neuronas dopaminérgicas que sustenta los síntomas del Parkinson. Aunque esta mutación en particular es rara, los mismos temas—deficiencia lisosomal, eliminación proteica deficiente y sobrecarga de alfa-sinucleína—aparecen también en la enfermedad de Parkinson más común y no familiar. Por tanto, el trabajo refuerza la idea de que restaurar los sistemas de limpieza del cerebro, por ejemplo mediante fármacos que activen TFEB o estimulen la autofagia como la rapamicina o sucesores más seguros, podría ser una estrategia prometedora para ralentizar o prevenir la neurodegeneración relacionada con el Parkinson.
Cita: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Palabras clave: Enfermedad de Parkinson, lisosomas, alfa-sinucleína, neuronas dopaminérgicas, rapamicina