Clear Sky Science · pl
Mutant skrócony PARK19 Dnajc6 powoduje niedobór lizosomalny prowadzący do podwyższenia patologicznego α-synukleiny i neurodegeneracji komórek dopaminergicznych istoty czarnej u myszy knockin PARK19
Dlaczego to badanie na myszach ma znaczenie dla choroby Parkinsona
Choroba Parkinsona odbiera ludziom zdolność ruchu, stopniowo zabijając niewielką grupę komórek mózgowych, które wytwarzają przekaźnik chemiczny — dopaminę. W większości przypadków choroba wynika z mieszanki wieku i czynników środowiskowych, ale u części chorych przyczyną są defekty pojedynczych genów, które stanowią cenne wskazówki co do mechanizmów choroby. To badanie analizuje jeden z takich genów, DNAJC6, wykorzystując specjalnie zmodyfikowaną mysz, aby pokazać, jak drobna zmiana wewnątrz komórek mózgowych może sparaliżować ich systemy utylizacji odpadów, przeładować je lepkim białkiem zwanym alfa‑synukleiną i w efekcie doprowadzić do utraty neuronów oraz objawów przypominających Parkinsona. 
Uszkodzony białkowy pomocnik w komórkach mózgu
Gen DNAJC6 koduje białko występujące niemal wyłącznie w neuronach, gdzie pomaga zarządzać klatryną — otoczkowatą cząsteczką, która formuje i recyklinguje błony komórkowe. W kilku rodzinach z wczesnym początkiem choroby Parkinsona znanej jako PARK19 obie kopie DNAJC6 noszą uszkadzające zmiany, które odcinają krytyczny koniec białka. Aby naśladować tę sytuację, badacze użyli edycji genów CRISPR, tworząc myszy, których wersja genu (Dnajc6 u myszy) kończy się wcześniej w równoważnym miejscu. Homozygotyczne myszy „Q787X” wytwarzają tylko skróconą formę białka, podczas gdy heterozygotyczne mają jedną normalną i jedną mutantową kopię, podobnie jak zdrowi nosiciele w ludzkich rodzinach.
Od wadliwego genu do problemów z ruchem
Zespół obserwował myszy w miarę starzenia się i testował ich zdolności motoryczne. W wieku pięciu miesięcy wszystkie grupy zachowywały się normalnie. Jednak do dziewiątego miesiąca myszy z dwiema kopiami mutanta rozwinęły klasyczne problemy motoryczne przypominające Parkinsona: poruszały się wolniej, pokonywały krótsze dystanse i potrzebowały więcej czasu, by zejść po słupku testowym — co odpowiada hipokinezji i bradykinezji u pacjentów. Po zbadaniu mózgów zaobserwowano wyraźną utratę neuronów produkujących dopaminę w istocie czarnej oraz przerzedzenie ich zakończeń nerwowych w prążkowiu. Te wrażliwe neurony zawierały też skupiska fosforylowanej alfa‑synukleiny, zbliżone do patologii ciałek Lewy’ego obserwowanej w ludzkiej chorobie Parkinsona. Myszom heterozygotycznym, mającym tylko jedną zmutowaną kopię, te zmiany się nie pojawiły.
Gdy komórkowy recykling zawodzi
Aby ustalić łańcuch zdarzeń, autorzy przyjrzeli się ośrodkom recyklingu komórkowego: lizosomom. W neuronach dopaminowych z myszy mutantów poziomy ciężkiego łańcucha klatryny były obniżone, co prawdopodobnie zaburza proces zwany autofagiczną odnową lizosomów — sposób, w jaki komórki odnawiają pulę lizosomów. Rzeczywiście liczba lizosomów spadła, a markery upośledzonej „samozjadliwości” komórkowej (autofagii) wzrosły. Co istotne, kluczowy enzym lizosomalny, katepsyna D, zmalał, podczas gdy alfa‑synukleina oraz jej bardziej toksyczne formy — fosforylowane i oligomeryczne — gromadziły się i stawały się nawet oporne na trawienie. Ta lizosomalna porażka była selektywna: sąsiednie neurony hamujące w innych rejonach nie wykazywały tych samych deficytów, co podkreśla szczególną wrażliwość komórek dopaminergicznych istoty czarnej. 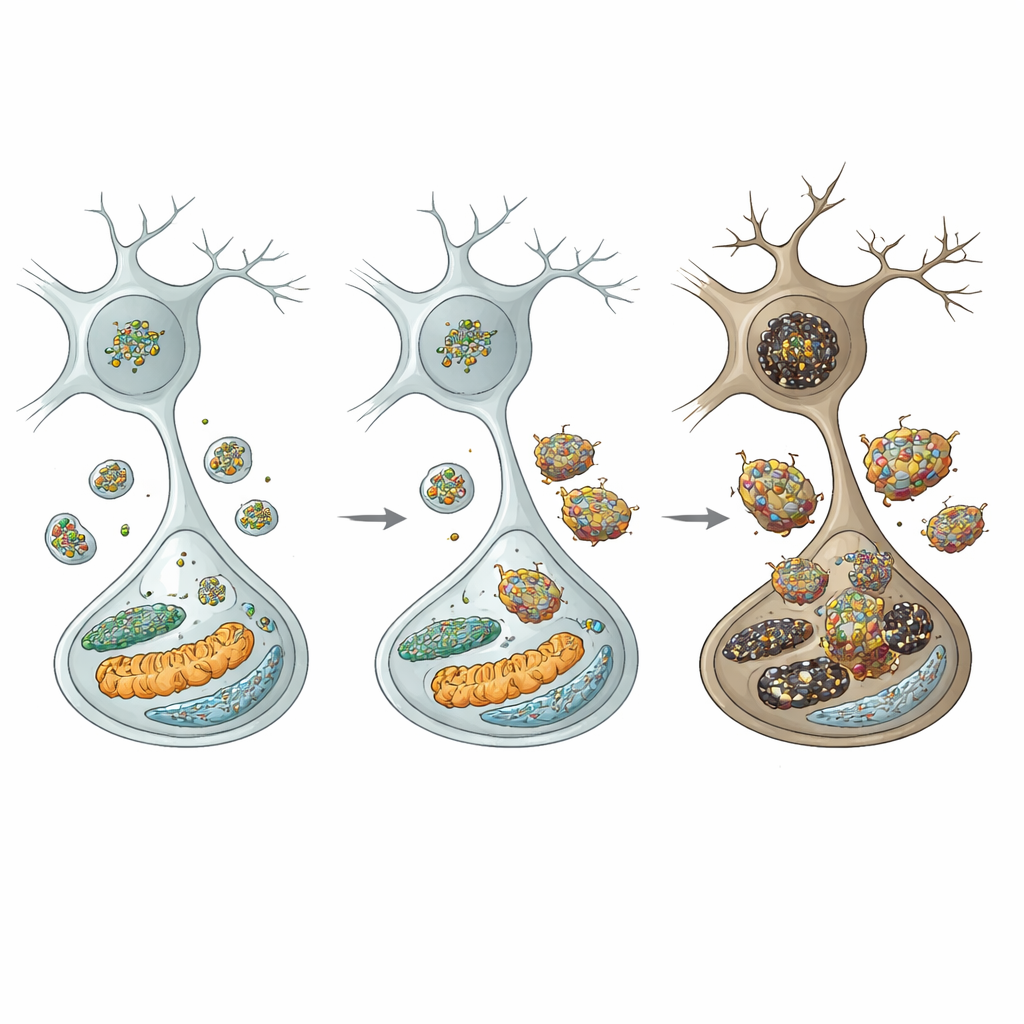
Rozprzestrzenianie się uszkodzeń wewnątrz i poza neuronem
Nadmierna alfa‑synukleina nie pozostawała w jednym miejscu. Gromadziła się na dwóch istotnych strukturach wewnętrznych — siateczce śródplazmatycznej i mitochondriach — gdzie wywoływała stres i uszkodzenia. W siateczce śródplazmatycznej uruchamiała odpowiedzi na nieprawidłowo sfałdowane białka i sygnały sprzyjające śmierci komórki. W mitochondriach zmniejszała aktywność kompleksu I odpowiedzialnego za produkcję energii, zwiększała poziom reaktywnych form tlenu i promowała uwalnianie cytochromu c, co aktywuje enzymy „samobójcze” zwane kaspazami. Równocześnie oligomeryczna alfa‑synukleina prawdopodobnie przeciekała z neuronów i pobudzała pobliskie mikrogleje, strażników immunologicznych mózgu. Mikrogleje przestawiały się w stan prozapalny, uruchamiały kompleks zapalny NLRP3 i uwalniały cząsteczki zapalne, takie jak IL‑1β, IL‑18 i TNF‑α. Te sygnały z kolei aktywowały dodatkowe ścieżki śmierci w neuronach dopaminowych, w tym zależne od JNK i necroptozy, tworząc błędne koło uszkodzeń.
Lek, który przywraca komórkową załogę sprzątającą
Następnie badacze sprawdzili, czy zwiększenie produkcji lizosomów może przerwać ten kaskadę. Leczyli młode myszy mutantów rapamycyną, lekiem, który pośrednio uwalnia główny przełącznik TFEB, pozwalając mu wejść do jądra i uruchomić geny budujące lizosomy, jednocześnie stymulując autofagię. Po czterech miesiącach myszy mutanty leczone rapamycyną odzyskały poziomy markerów lizosomalnych i katepsyny D w istocie czarnej, a nagromadzenie alfa‑synukleiny było znacznie mniejsze. Co niezwykłe, te myszy były w dużej mierze chronione przed utratą neuronów dopaminowych zarówno w istocie czarnej, jak i w sąsiednim polu brzusznym nakrywki, a ich wydolność ruchowa poprawiła się w kierunku normy. Działając na wysokim poziomie w sieci obsługi odpadów komórkowych, rapamycyna skutecznie stłumiła dalszy kaskadowy efekt gromadzenia białka, stresu organelli, zapalenia i śmierci komórek.
Co to oznacza dla rozumienia i leczenia Parkinsona
Krótko mówiąc, praca ta pokazuje, jak pojedynczy odziedziczony defekt w neuron‑specyficznym białku pomocniczym może wywołać efekt domina: mniej lizosomów, osłabiona aktywność katepsyny D, narastające poziomy toksycznej alfa‑synukleiny, uszkodzenia kluczowych przedziałów komórkowych, nadmierna reakcja zapalna i w końcu utrata neuronów dopaminowych leżąca u podstaw objawów Parkinsona. Chociaż ta konkretna mutacja jest rzadka, te same motywy — niedobór lizosomalny, słabe usuwanie białek i przeładowanie alfa‑synukleiną — pojawiają się również w częstszych, niefamilijnych postaciach choroby Parkinsona. Praca wzmacnia zatem ideę, że przywrócenie systemów sprzątania mózgu, na przykład przez aktywację TFEB lub zwiększenie autofagii lekami takimi jak rapamycyna lub bezpieczniejsze jej następcy, może być obiecującą strategią spowalniania lub zapobiegania związanej z Parkinsonem neurodegeneracji.
Cytowanie: Wang, HL., Chen, YL., Chiu, TJ. et al. PARK19 truncation mutant Dnajc6 causes lysosomal deficiency-induced upregulation of pathologic α-synuclein and neurodegeneration of substantia nigra dopaminergic cells in PARK19 knockin mice. npj Parkinsons Dis. 12, 102 (2026). https://doi.org/10.1038/s41531-026-01317-8
Słowa kluczowe: choroba Parkinsona, lizosomy, alfa-synukleina, neurony dopaminergiczne, rapamycyna