Clear Sky Science · zh
通过抑制非典型的 Akt–mTORC1 激活,在多种同基因系模型中拯救 GNE 肌病的自噬缺陷
为何肌肉自我清理至关重要
我们的肌肉不断分解损耗部件并重建它们。这种内部的“家务”——细胞回收(自噬)——使肌纤维在一生中保持强健。在一种称为 GNE 肌病的罕见遗传病中,青年人会逐渐失去肌力,但从单一基因缺陷到肌肉萎缩的事件链一直不清楚。本研究逐步追踪了这条链路,使用干细胞模型、基因工程小鼠肌细胞和微型神经肌器官,展示了肌细胞周围受扰动的环境如何关闭细胞内的回收机制,以及现有的抗癌药物如何可能帮助重新启动该机制。
从基因改变到肌无力
GNE 肌病由一种基因的变异引起,该基因通常帮助细胞合成唾液酸——一种修饰许多表面分子的小糖。当该基因失效时,肌纤维缺乏唾液酸,最终在患者活检中出现边缘空泡(rimmed vacuoles)等显微结构。早期研究暗示自噬受损可能是问题的一部分,但从基因缺陷到自噬失败的具体连线尚不明确。作者首先比较了来源于人类干细胞的肌细胞中基因活动,这些细胞唯一不同的是是否携带常见的 GNE 突变。他们观察到与回收相关的基因集合发生了广泛变化,表明在病变细胞中维持细胞清理的通路持续受扰。
构建配对的细胞模型
为深入研究机制,团队在一株标准小鼠肌母细胞系中敲除了 Gne 基因,构建了更简单的肌细胞系统。这些工程化细胞表现出与人类模型和患者相同的唾液酸缺乏。在常规生长条件下它们看起来相对正常,但当撤去糖类以模拟代谢应激时,敲除细胞更容易死亡。详尽的蛋白测量和成像显示,在这些受压细胞中,被称为自噬小体的回收结构形成不良,物质进入细胞废物隔间的正常流动受阻。这确认了当 GNE 缺陷肌肉在能量需求下受到挑战时,自噬失败是一个核心弱点。
细胞外如何堵塞细胞内
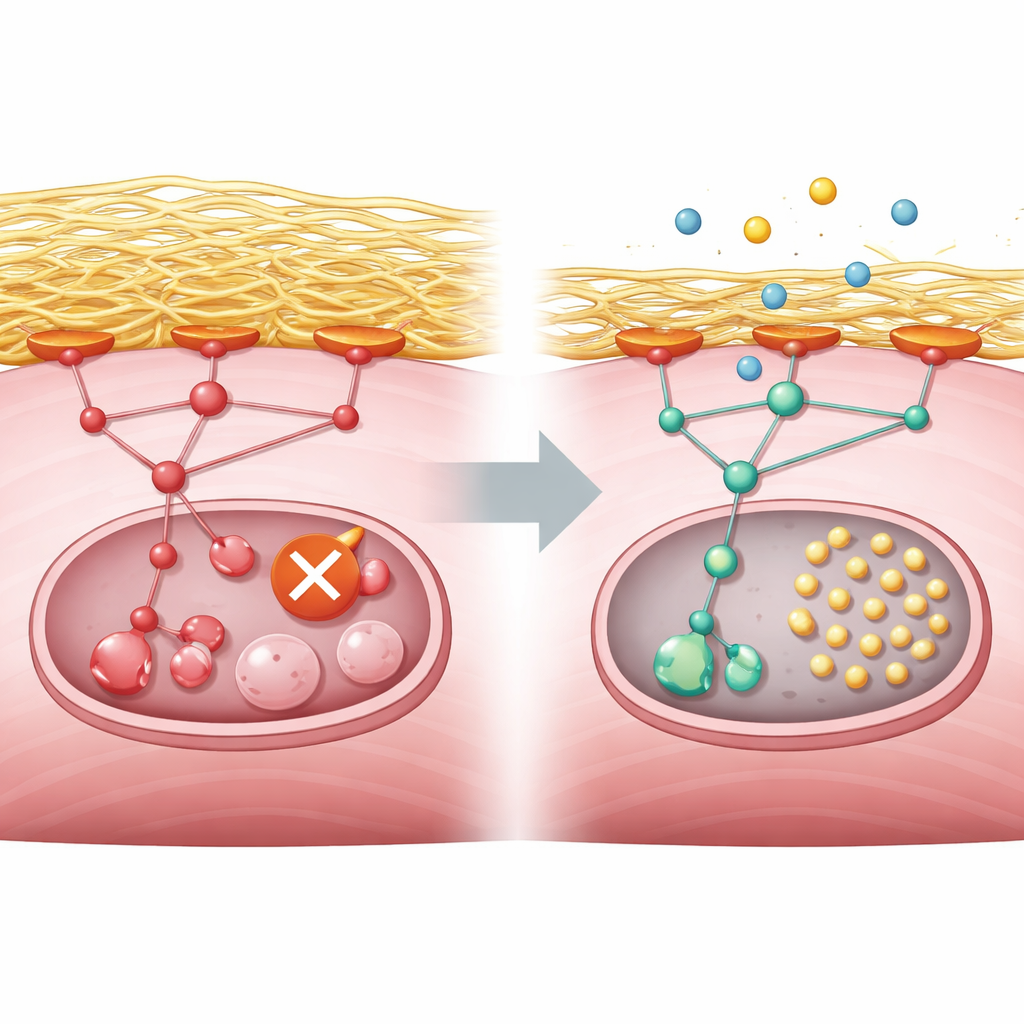
研究者接着询问在分子水平上是什么阻断了自噬。他们的分析指向由 PI3K、AKT 和 mTORC1 蛋白驱动的过度活跃的生长控制通路,这些通路通常对营养和生长信号作出反应。在健康肌肉中,能量短缺会抑制该通路,从而使启动酶 ULK1 激活并触发自噬。然而在 GNE 缺陷细胞中,即使在饥饿条件下该通路也保持开启。与此同时,细胞过度产生胶原和其他细胞外基质成分。这种过量的基质增强了细胞表面与环境之间的接触点,进而激活一个称为粘着斑激酶(focal adhesion kinase)的中继分子,并将信号传入 PI3K–AKT–mTORC1 通路。结果是 ULK1 上持续存在的化学抑制标记,使其处于关闭状态,阻止自噬的启动。
测试一种可放松刹车的药物
根据该连线图,团队在一个大型药物诱导基因表达数据库中搜索与疾病特征相反效应的药物。许多顶级候选是 PI3K 或 mTOR 抑制剂,研究组选择了 copanlisib——一种获批用于某些淋巴瘤的静脉给药 PI3K 抑制剂——进行进一步测试。在 GNE 敲除的小鼠肌细胞中,copanlisib 降低了过度活跃的通路信号,去除了 ULK1 的抑制性标记并恢复了回收活性,尽管它并未修复原始的唾液酸缺乏。随后作者将研究转向由携带 GNE 突变的人类干细胞长出的三维神经肌器官。这些微型神经肌单元再现了低唾液酸水平、自噬缺陷和 mTORC1 过度活性。在营养应激期间用 copanlisib 处理病变器官时,肌纤维内形成了更多的自噬点,表明细胞清理功能得到恢复。
这对 GNE 肌病患者意味着什么
简而言之,这项工作将肌细胞中一个合成糖类的故障基因与细胞外基质的过度积累联系起来,后者过度刺激生长控制通路并关闭了细胞的清理机制。研究显示,这一模式在工程化小鼠细胞、人类干细胞衍生肌肉和器官以及患者活检的基因表达中均有出现。虽然 copanlisib 尚未成为 GNE 肌病的治疗方法,但这些实验表明,谨慎下调 PI3K–AKT–mTORC1 通路可能解除自噬阻滞,有望减缓肌肉损伤。未来的临床研究需要评估这种方法在人群中的安全性与疗效,但该工作为治疗提供了明确且可检验的靶点。
引用: Kim, DW., Kwon, EJ., Kwon, H. et al. Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models. Exp Mol Med 58, 1187–1202 (2026). https://doi.org/10.1038/s12276-026-01701-7
关键词: GNE 肌病, 肌肉自噬, 细胞外基质, AKT mTORC1 通路, copanlisib