Clear Sky Science · pt
Autofagia defeituosa na miopatia por GNE é resgatada pela inibição da ativação não canônica Akt–mTORC1 em vários modelos isogênicos
Por que a autolimpeza muscular importa
Nossos músculos estão constantemente degradando componentes desgastados e reconstruindo-os. Essa manutenção interna, chamada reciclagem celular, mantém as fibras musculares fortes ao longo da vida. Em uma condição hereditária rara conhecida como miopatia por GNE, adultos jovens perdem força muscular de forma lenta, contudo a sequência de eventos que parte de um único gene defeituoso até a perda muscular não estava clara. Este estudo segue essa sequência passo a passo, usando modelos de células-tronco, células musculares de camundongo geneticamente modificadas e organoides neuromusculares em miniatura para mostrar como um ambiente ao redor da célula muscular perturbado pode desligar a reciclagem dentro da célula e como um fármaco já usado contra câncer pode ajudar a religá-la.
Da alteração genética aos músculos fracos
A miopatia por GNE é causada por variações em um gene que normalmente auxilia as células a fabricar ácido siálico, um pequeno açúcar que adorna muitas moléculas de superfície. Quando o gene falha, as fibras musculares ficam com deficiência de ácido siálico e eventualmente desenvolvem vacúolos com borda, estruturas semelhantes a bolhas vistas ao microscópio em biópsias de pacientes. Trabalhos anteriores sugeriram que a autofagia defeituosa poderia ser parte do problema, mas a ligação exata do defeito genético à falha na reciclagem era desconhecida. Os autores começaram comparando a atividade gênica em células musculares derivadas de células-tronco humanas que diferiam apenas por carregarem ou não uma mutação comum no GNE. Eles observaram mudanças amplas em conjuntos de genes relacionados à reciclagem, sugerindo que as vias de manutenção estavam consistentemente perturbadas nas células doentes.
Construindo modelos celulares correspondentes
Para aprofundar os mecanismos, a equipe criou um sistema celular muscular mais simples deletando o gene Gne em uma linha padrão de mióblastos de camundongo. Essas células modificadas exibiram a mesma falta de ácido siálico vista nos modelos humanos e em pacientes. Em condições de crescimento rotineiras pareciam relativamente normais, mas quando os suprimentos de açúcar foram retirados para mimetizar estresse metabólico, as células knockout eram muito mais propensas a morrer. Medições detalhadas de proteínas e imagens revelaram que, nessas células estressadas, as estruturas de reciclagem chamadas autofagossomos se formavam de maneira deficiente e o fluxo normal de material para compartimentos de degradação celular estava reduzido. Isso confirmou que a falha na reciclagem é uma fragilidade central revelada quando o músculo deficiente em GNE é pressionado pela demanda de energia.
Como o exterior da célula emperra o interior
Os pesquisadores perguntaram em seguida o que estava bloqueando a reciclagem a nível molecular. Suas análises apontaram para uma via de controle de crescimento hiperativada, conduzida por proteínas conhecidas como PI3K, AKT e mTORC1, que normalmente respondem a nutrientes e sinais de crescimento. No músculo saudável, a falta de energia silencia essa via e permite que uma enzima inicial chamada ULK1 dispare a reciclagem. Nas células deficientes em GNE, porém, a via permanecia ativada mesmo durante a privação de nutrientes. Ao mesmo tempo, as células superproduziam colágeno e outros componentes do arcabouço ao redor, a matriz extracelular. Esse excesso de matriz fortaleceu pontos de contato entre a superfície celular e seu entorno, o que por sua vez ativou uma molécula de retransmissão chamada quinase de adesão focal e alimentou a via PI3K–AKT–mTORC1. O resultado foi uma marca química persistente na ULK1 que a mantém em estado inativo, impedindo que a reciclagem sequer comece.
Testando um fármaco que afrouxa o freio
Munidos desse diagrama de conexões, a equipe buscou em um grande banco de dados de alterações gênicas induzidas por medicamentos para achar compostos cujos efeitos fossem opostos à assinatura da doença. Muitos dos principais candidatos eram inibidores de PI3K ou mTOR, e eles selecionaram o copanlisibe, um inibidor de PI3K intravenoso aprovado para certos linfomas, para testes adicionais. Em células musculares de camundongo com knockout de Gne, o copanlisibe reduziu o sinal excessivo da via, removeu a marca inibitória da ULK1 e restaurou a atividade de reciclagem, embora não consertasse a falta original de ácido siálico. Os autores então passaram a organoides neuromusculares tridimensionais cultivados a partir de células-tronco humanas portadoras da mutação em GNE. Essas unidades musculoneurais em miniatura reproduziram níveis baixos de ácido siálico, defeitos na reciclagem e hiperativação de mTORC1. Quando tratadas com copanlisibe durante o estresse nutricional, os organoides doentes formaram muito mais pontos de reciclagem dentro das fibras musculares, indicando a recuperação da manutenção celular.
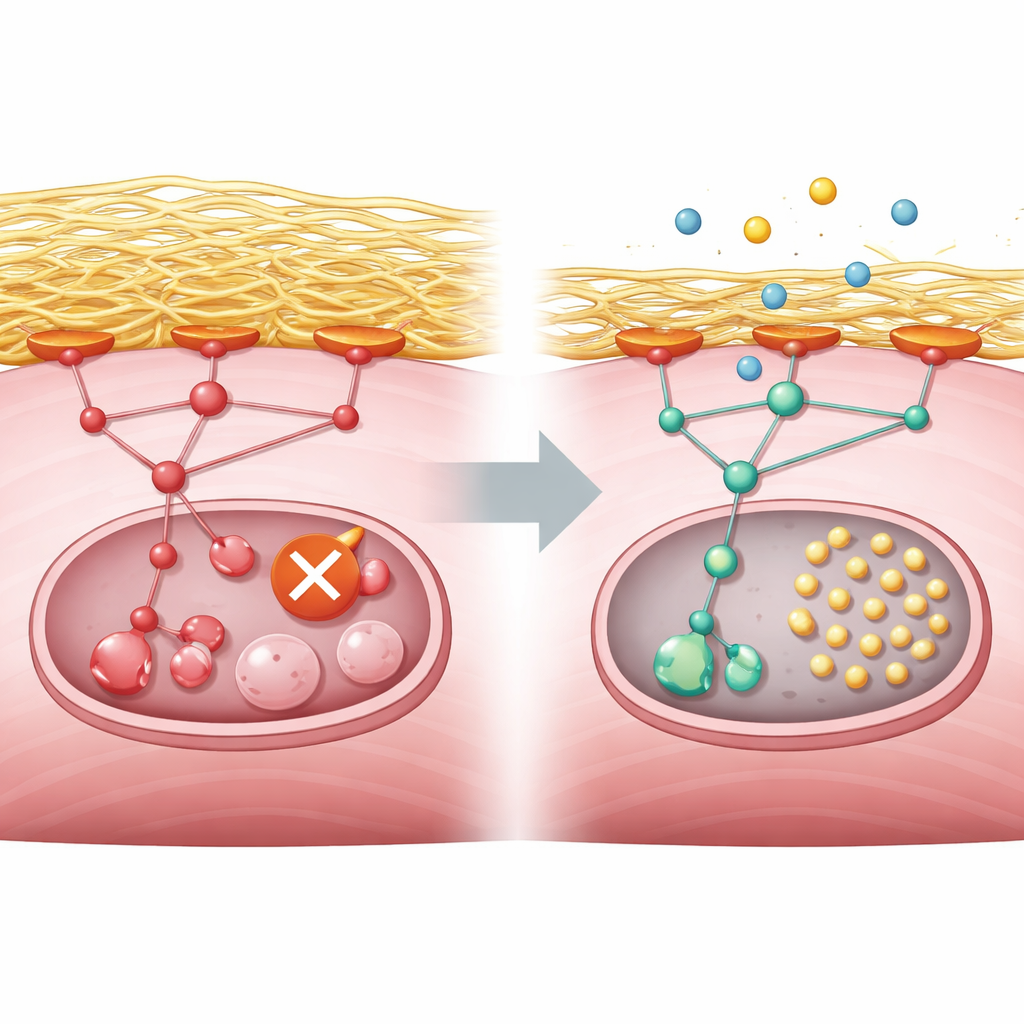
O que isso significa para pessoas com miopatia por GNE
Em termos simples, este trabalho liga um gene defeituoso de síntese de açúcar nas células musculares ao acúmulo excessivo do arcabouço ao redor, que hiperestimula uma via de controle de crescimento e desliga a equipe de limpeza da célula. O estudo mostra que esse mesmo padrão aparece em células de camundongo modificadas, em músculo derivado de células-tronco humanas e em organoides, bem como na atividade gênica de biópsias de pacientes. Embora o copanlisibe ainda não seja um tratamento para a miopatia por GNE, os experimentos sugerem que reduzir com cuidado a via PI3K–AKT–mTORC1 pode aliviar o bloqueio da reciclagem e potencialmente retardar o dano muscular. Estudos clínicos futuros precisarão determinar se essa abordagem é segura e eficaz em pessoas, mas o trabalho fornece um alvo claro e testável para terapia.
Citação: Kim, DW., Kwon, EJ., Kwon, H. et al. Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models. Exp Mol Med 58, 1187–1202 (2026). https://doi.org/10.1038/s12276-026-01701-7
Palavras-chave: miopatia por GNE, autofagia muscular, matriz extracelular, via AKT mTORC1, copanlisibe