Clear Sky Science · en
Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models
Why muscle self cleaning matters
Our muscles constantly break down worn out parts and rebuild them. This internal housekeeping, called cellular recycling, keeps muscle fibers strong over a lifetime. In a rare inherited condition known as GNE myopathy, young adults slowly lose muscle strength, yet the chain of events leading from a single faulty gene to wasting muscle has been unclear. This study follows that chain step by step, using stem cell models, engineered mouse muscle cells and miniature neuromuscular organoids to show how a disrupted environment around the muscle cell can switch off recycling inside the cell and how an existing cancer drug might help switch it back on.
From gene change to weak muscles
GNE myopathy is caused by changes in a gene that normally helps cells make sialic acid, a small sugar that decorates many surface molecules. When the gene fails, muscle fibers become undersupplied with sialic acid and eventually develop rimmed vacuoles, bubble like structures seen under the microscope in patient biopsies. Earlier work hinted that defective cellular recycling could be part of the problem, but the precise wiring from gene defect to failed recycling was unknown. The authors began by comparing gene activity in muscle cells grown from human stem cells that differed only in whether they carried a common GNE mutation. They saw broad shifts in recycling related gene sets, suggesting that housekeeping pathways were consistently disturbed in diseased cells.
Building matching cell models
To dig into the mechanisms, the team created a simpler muscle cell system by deleting the Gne gene in a standard mouse myoblast line. These engineered cells showed the same lack of sialic acid seen in human models and in patients. Under routine growth conditions they looked fairly normal, but when sugar supplies were withdrawn to mimic metabolic stress, the knockout cells were much more likely to die. Detailed protein measurements and imaging revealed that, in these stressed cells, recycling structures called autophagosomes formed poorly and the normal flow of material into cellular waste compartments was blunted. This confirmed that recycling failure is a core weakness revealed when GNE deficient muscle is pushed by energy demand.
How the outside of the cell jams the inside
The researchers next asked what was blocking recycling at the molecular level. Their analyses pointed to an overactive growth control pathway driven by proteins known as PI3K, AKT and mTORC1, which normally respond to nutrients and growth signals. In healthy muscle, energy shortage quiets this pathway and allows a starter enzyme called ULK1 to trigger recycling. In GNE deficient cells, however, the pathway stayed switched on even during starvation. At the same time, the cells overproduced collagen and other components of the surrounding scaffold, or extracellular matrix. This excess scaffold strengthened contact points between the cell surface and its environment, which in turn activated a relay molecule called focal adhesion kinase and fed into the PI3K–AKT–mTORC1 pathway. The result was a persistent chemical tag on ULK1 that keeps it in an off state, preventing recycling from even starting.
Testing a drug that loosens the brake
Armed with this wiring diagram, the team searched a large database of drug induced gene changes to find medicines whose effects ran opposite to the disease signature. Many of the top hits were blockers of PI3K or mTOR, and they selected copanlisib, an approved intravenous PI3K inhibitor used for certain lymphomas, for further tests. In GNE knockout mouse muscle cells, copanlisib reduced the overactive pathway signal, removed the inhibitory tag from ULK1 and restored recycling activity, even though it did not repair the original sialic acid shortage. The authors then turned to three dimensional neuromuscular organoids grown from human stem cells carrying the GNE mutation. These miniature muscle nerve units reproduced low sialic acid levels, recycling defects and mTORC1 overactivity. When treated with copanlisib during nutrient stress, diseased organoids formed many more recycling puncta inside muscle fibers, indicating revived housekeeping.
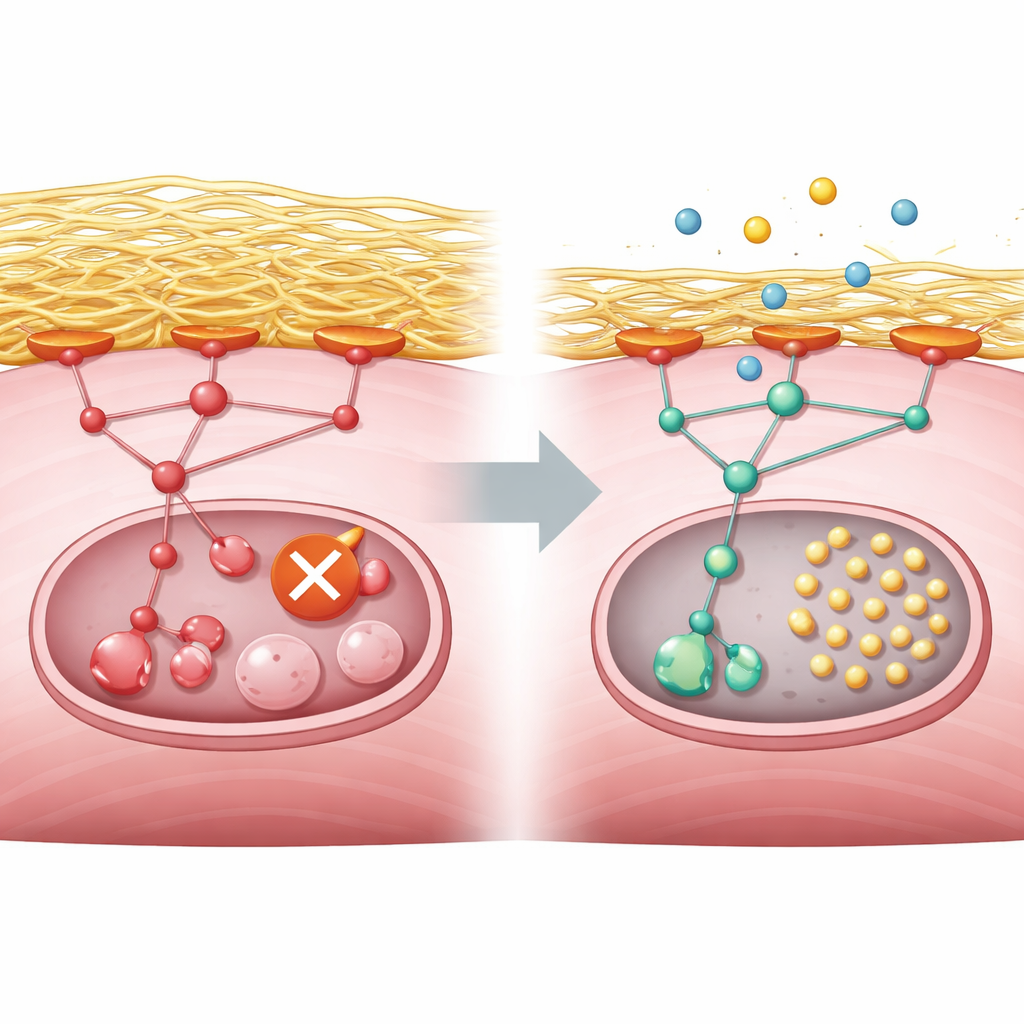
What this means for people with GNE myopathy
Put simply, this work links a faulty sugar making gene in muscle cells to excessive buildup of surrounding scaffold, which overstimulates a growth control pathway and shuts down the cell’s cleanup crew. The study shows that this same pattern appears in engineered mouse cells, human stem cell derived muscle and organoids, and in gene activity from patient biopsies. While copanlisib is not yet a treatment for GNE myopathy, the experiments suggest that carefully dialing down the PI3K–AKT–mTORC1 pathway could relieve the recycling block and potentially slow muscle damage. Future clinical studies will need to determine whether this approach is safe and effective in people, but the work provides a clear, testable target for therapy.
Citation: Kim, DW., Kwon, EJ., Kwon, H. et al. Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models. Exp Mol Med 58, 1187–1202 (2026). https://doi.org/10.1038/s12276-026-01701-7
Keywords: GNE myopathy, muscle autophagy, extracellular matrix, AKT mTORC1 pathway, copanlisib