Clear Sky Science · pl
Defekt autofagii w miopatii GNE jest naprawiany przez hamowanie niekanonicznej aktywacji Akt–mTORC1 w wielu izogenicznych modelach
Dlaczego samopielęgnacja mięśni ma znaczenie
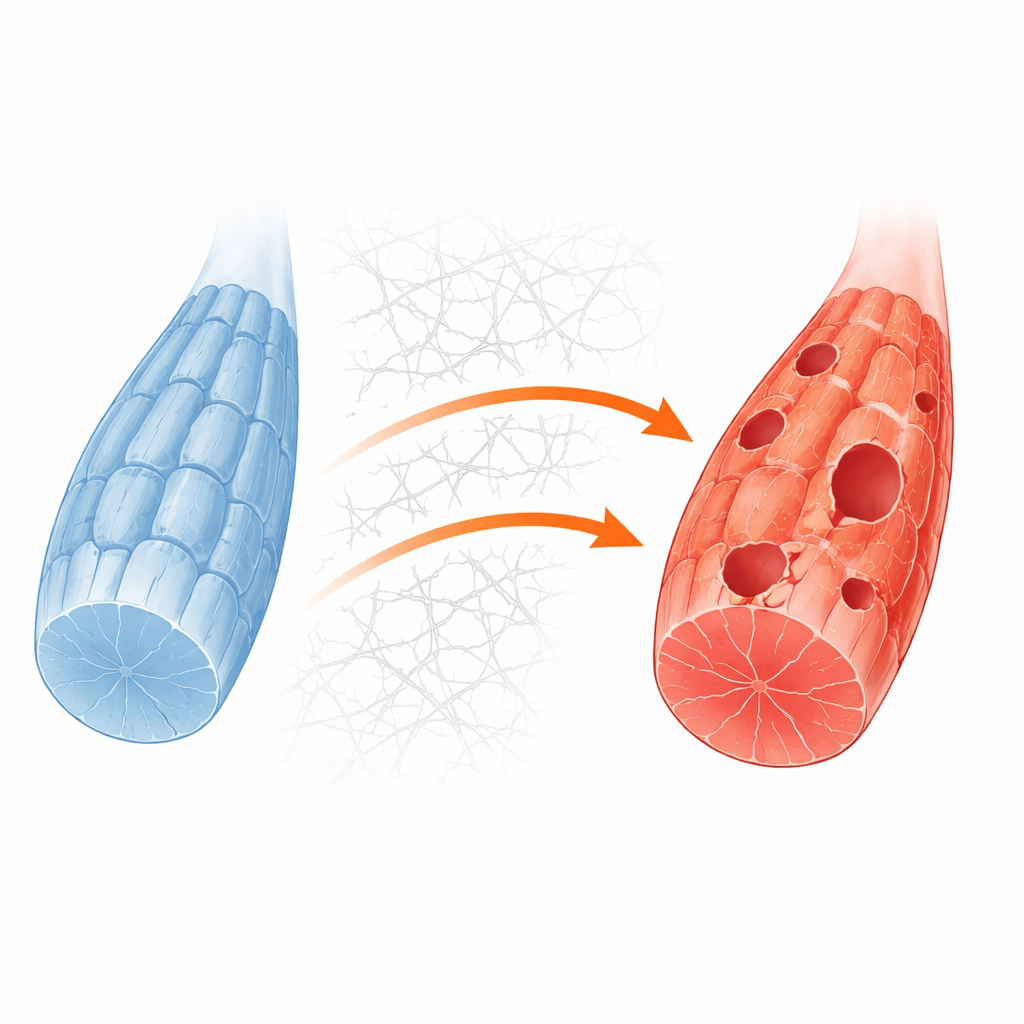
Nasze mięśnie nieustannie rozkładają zużyte elementy i je odbudowują. Ta wewnętrzna gospodarka, zwana recyklingiem komórkowym, utrzymuje włókna mięśniowe w dobrej kondycji przez całe życie. W rzadkiej chorobie dziedzicznej znanej jako miopatia GNE młodzi dorośli stopniowo tracą siłę mięśni, jednak łańcuch zdarzeń prowadzących od jednej uszkodzonej kopii genu do zanikającego mięśnia pozostawał niejasny. W tym badaniu śledzono ten łańcuch krok po kroku, wykorzystując modele z komórek macierzystych, zmodyfikowane mysie komórki mięśniowe i miniaturowe organoidy nerwowo‑mięśniowe, aby pokazać, jak zaburzone otoczenie komórki mięśniowej może wyłączyć recykling wewnątrz komórki i jak istniejący lek przeciwnowotworowy może pomóc go przywrócić.
Od zmiany w genie do słabych mięśni
Miopatia GNE jest spowodowana zmianami w genie, który normalnie pomaga komórkom w syntezie kwasu sjalowego, małego cukru występującego na wielu cząsteczkach powierzchniowych. Gdy gen zawodzi, włókna mięśniowe stają się niedostatecznie wysialowane i ostatecznie rozwijają się w nich tzw. rimmed vacuoles — pęcherzykowate struktury widoczne w biopsjach pacjentów. Wcześniejsze prace sugerowały, że defekt recyklingu komórkowego może być częścią problemu, ale dokładne powiązanie pomiędzy usterką genetyczną a niewydolnością recyklingu pozostawało nieznane. Autorzy zaczęli od porównania aktywności genów w komórkach mięśniowych wyhodowanych z ludzkich komórek macierzystych różniących się tylko obecnością powszechnej mutacji GNE. Zaobserwowali szerokie przesunięcia w zestawach genów związanych z recyklingiem, co sugerowało, że szlaki porządkowania komórkowego są konsekwentnie zaburzone w komórkach chorych.
Budowanie dopasowanych modeli komórkowych
Aby zagłębić się w mechanizmy, zespół stworzył prostszy system komórek mięśniowych, usuwając gen Gne w standardowej linii mysich mioblastów. Te zmodyfikowane komórki wykazały ten sam niedobór kwasu sjalowego, co modele ludzkie i pacjenci. W rutynowych warunkach wzrostu wyglądały stosunkowo normalnie, ale gdy odjęto dostawy cukru, aby naśladować stres metaboliczny, komórki z wyciętym genem znacznie częściej umierały. Szczegółowe pomiary białek i obrazowanie wykazały, że w tych zestresowanych komórkach struktury recyklingowe zwane autofagosomami formowały się słabo, a normalny przepływ materiału do komórkowych przekaźników degradacyjnych był zahamowany. Potwierdziło to, że niewydolność recyklingu jest kluczową słabością ujawnianą, gdy mięśnie pozbawione GNE są obciążone zapotrzebowaniem energetycznym.
Jak zewnętrze komórki zatyka wnętrze
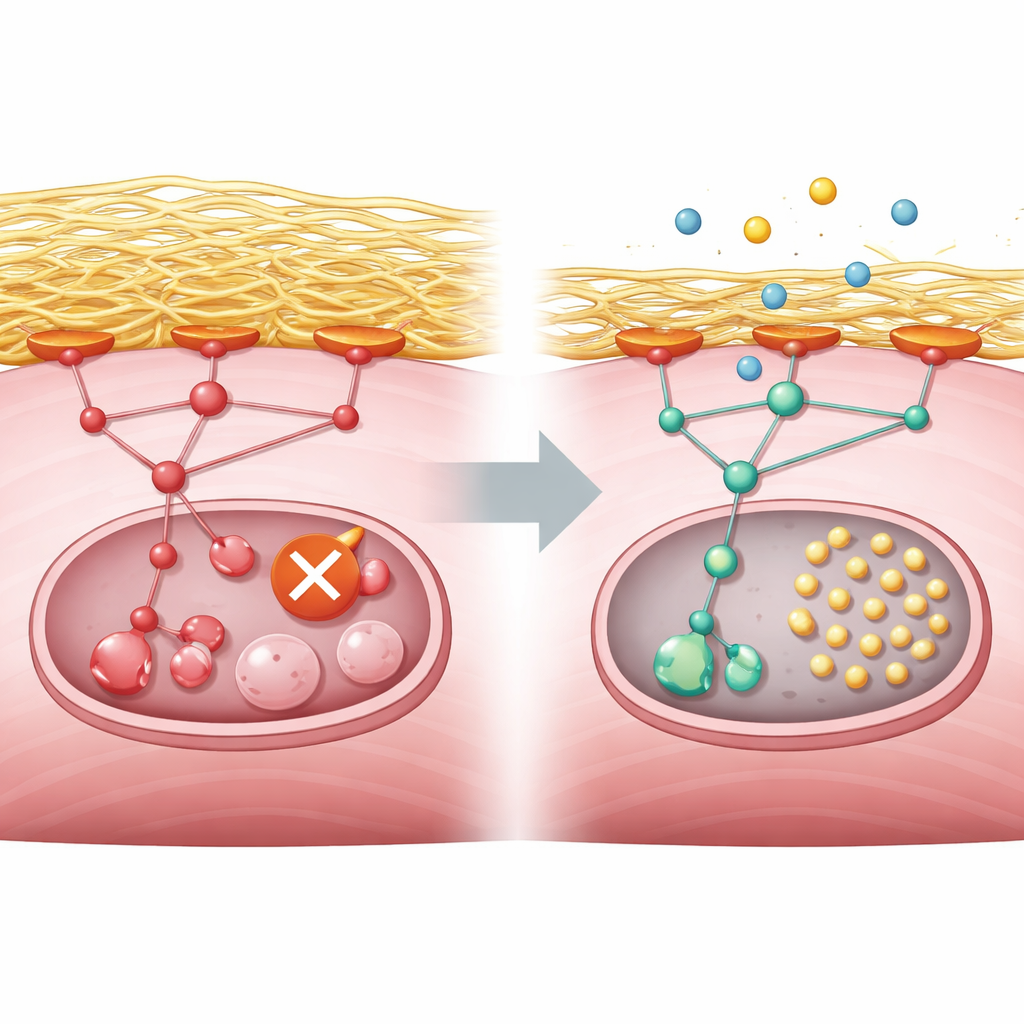
Naukowcy następnie zapytali, co na poziomie molekularnym hamuje recykling. Ich analizy wskazały na nadaktywowany szlak kontroli wzrostu napędzany przez białka znane jako PI3K, AKT i mTORC1, które normalnie reagują na dostępność składników odżywczych i sygnały wzrostu. W zdrowym mięśniu niedobór energii wycisza ten szlak i pozwala enzymowi startowemu ULK1 uruchomić recykling. W komórkach pozbawionych GNE natomiast szlak pozostawał aktywny nawet podczas głodzenia. Jednocześnie komórki nadmiernie produkowały kolagen i inne składniki otaczającego rusztowania, czyli macierzy zewnątrzkomórkowej. Ten nadmiar wzmacniał punkty kontaktu między powierzchnią komórki a jej środowiskiem, co z kolei aktywowało kinazę ognisk adhezji (focal adhesion kinase) i włączało sygnał do szlaku PI3K–AKT–mTORC1. Wynikiem był utrzymujący się chemiczny znacznik na ULK1, który pozostawia go w stanie wyłączonym, uniemożliwiającym rozpoczęcie recyklingu.
Testowanie leku, który luzuje hamulec
Mając ten schemat połączeń, zespół przeszukał dużą bazę danych zmian ekspresji genów indukowanych lekami, szukając substancji o efektach przeciwnych do sygnatury choroby. Wiele z najlepszych trafień stanowiły inhibitory PI3K lub mTOR, a do dalszych testów wybrano copanlisib — zatwierdzony dożylny inhibitor PI3K stosowany w niektórych chłoniakach. W mysich komórkach mięśniowych z wyciętym Gne copanlisib zmniejszył nadmierny sygnał szlaku, usunął hamujący znacznik z ULK1 i przywrócił aktywność recyklingu, mimo że nie naprawiał pierwotnego niedoboru kwasu sjalowego. Autorzy przeszli następnie do trójwymiarowych organoidów nerwowo‑mięśniowych wyhodowanych z ludzkich komórek macierzystych niosących mutację GNE. Te miniaturowe jednostki mięśniowo‑nerwowe odzwierciedlały niskie poziomy kwasu sjalowego, defekty recyklingu i nadaktywność mTORC1. Po leczeniu copanlisibem podczas niedoboru składników odżywczych chore organoidy tworzyły znacznie więcej punktów recyklingu wewnątrz włókien mięśniowych, co wskazywało na odnowioną wewnątrzkomórkową gospodarkę.
Co to oznacza dla osób z miopatią GNE
Krótko mówiąc, praca łączy wadliwy gen odpowiedzialny za syntezę cukru w komórkach mięśniowych z nadmiernym nagromadzeniem otaczającego rusztowania, które nadmiernie stymuluje szlak kontroli wzrostu i wyłącza komórkową ekipę sprzątającą. Badanie pokazuje, że ten sam schemat pojawia się w zmodyfikowanych komórkach mysich, mięśniach pochodzących z ludzkich komórek macierzystych, w organoidach oraz w aktywności genów z biopsji pacjentów. Chociaż copanlisib nie jest jeszcze leczeniem miopatii GNE, eksperymenty sugerują, że ostrożne stłumienie szlaku PI3K–AKT–mTORC1 mogłoby usunąć blokadę recyklingu i potencjalnie spowolnić uszkodzenia mięśni. Przyszłe badania kliniczne będą musiały ustalić, czy to podejście jest bezpieczne i skuteczne u ludzi, ale praca dostarcza jasnego, możliwego do przetestowania celu terapeutycznego.
Cytowanie: Kim, DW., Kwon, EJ., Kwon, H. et al. Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models. Exp Mol Med 58, 1187–1202 (2026). https://doi.org/10.1038/s12276-026-01701-7
Słowa kluczowe: miopatia GNE, autofagia mięśni, macierz zewnątrzkomórkowa, szlak AKT–mTORC1, copanlisib