Clear Sky Science · it
L’autofagia difettosa nella miopatia da GNE è ripristinata dall’inibizione dell’attivazione non canonica Akt–mTORC1 in più modelli isogenici
Perché la pulizia interna dei muscoli è importante
I nostri muscoli degradano costantemente le parti usurate e le ricostruiscono. Questo lavoro di manutenzione interna, chiamato riciclo cellulare, mantiene le fibre muscolari forti per tutta la vita. In una rara malattia ereditaria nota come miopatia da GNE, gli adulti giovani perdono gradualmente forza muscolare, ma la catena di eventi che porta da un singolo gene difettoso all’atrofia muscolare è rimasta poco chiara. Questo studio segue quella catena passo dopo passo, utilizzando modelli di cellule staminali, cellule muscolari murine ingegnerizzate e organoidi neuromuscolari in miniatura per mostrare come un ambiente alterato attorno alla cellula muscolare possa spegnere il riciclo all’interno della cellula e come un farmaco oncologico esistente potrebbe contribuire a riattivarlo.
Dal cambiamento genico ai muscoli deboli
La miopatia da GNE è causata da variazioni in un gene che normalmente aiuta le cellule a sintetizzare l’acido sialico, un piccolo zucchero che decorra molti elementi di superficie. Quando il gene non funziona, le fibre muscolari risultano carenti di acido sialico e alla fine sviluppano vacuoli con bordo, strutture a forma di bolla visibili al microscopio nelle biopsie dei pazienti. Studi precedenti avevano suggerito che un difetto nel riciclo cellulare potesse contribuire al problema, ma il collegamento preciso dal difetto genetico al fallimento del riciclo era sconosciuto. Gli autori hanno iniziato confrontando l’attività genica in cellule muscolari cresciute da cellule staminali umane che differivano solo per la presenza di una comune mutazione di GNE. Hanno osservato ampie alterazioni nei set genici legati al riciclo, suggerendo che i percorsi di manutenzione fossero sistematicamente disturbati nelle cellule malate.
Costruire modelli cellulari corrispondenti
Per approfondire i meccanismi, il gruppo ha creato un sistema cellulare muscolare più semplice eliminando il gene Gne in una linea standard di mioblasti murini. Queste cellule ingegnerizzate mostrarono la stessa carenza di acido sialico osservata nei modelli umani e nei pazienti. In condizioni di crescita ordinaria apparivano abbastanza normali, ma quando le riserve di zucchero venivano rimosse per mimare lo stress metabolico, le cellule knockout avevano una probabilità molto maggiore di morire. Misurazioni proteiche dettagliate e immagini rivelarono che, in queste cellule sotto stress, le strutture di riciclo chiamate autofagosomi si formavano male e il normale flusso di materiale verso i compartimenti di degradazione cellulare era ostacolato. Questo confermò che il fallimento del riciclo è una debolezza centrale che emerge quando il muscolo carente di GNE è spinto da un aumento della domanda energetica.
Come l’esterno della cellula inceppa l’interno
I ricercatori hanno quindi indagato cosa bloccasse il riciclo a livello molecolare. Le loro analisi indicarono un percorso di controllo della crescita iperattivato guidato da proteine note come PI3K, AKT e mTORC1, che normalmente rispondono a nutrienti e segnali di crescita. Nel muscolo sano, la carenza di energia silenzia questo circuito e permette a un enzima iniziale chiamato ULK1 di avviare il riciclo. Nelle cellule carenti di GNE, tuttavia, il percorso restava attivato anche durante la privazione di nutrienti. Allo stesso tempo, le cellule producevano in eccesso collagene e altri componenti della impalcatura circostante, la matrice extracellulare. Questo eccesso di impalcatura rafforzava i punti di contatto tra la superficie cellulare e l’ambiente circostante, attivando a sua volta una molecola di trasduzione chiamata chinasi delle adesioni focali (focal adhesion kinase) che alimentava la via PI3K–AKT–mTORC1. Il risultato era un marcatore chimico persistente su ULK1 che lo mantiene spento, impedendo l’avvio del riciclo.
Testare un farmaco che allenta il freno
Con questo schema operativo, il team ha cercato in un ampio database di cambiamenti genici indotti da farmaci per trovare medicine i cui effetti fossero opposti alla firma della malattia. Molte delle migliori corrispondenze erano inibitori di PI3K o mTOR, e hanno scelto il copanlisib, un inibitore IV della PI3K approvato per alcuni linfomi, per ulteriori test. Nelle cellule muscolari murine knockout per Gne, il copanlisib ridusse il segnale iperattivo del percorso, rimosse il marchio inibitorio da ULK1 e ripristinò l’attività di riciclo, anche se non riparò la carenza iniziale di acido sialico. Gli autori si sono poi rivolti a organoidi neuromuscolari tridimensionali coltivati da cellule staminali umane portatrici della mutazione GNE. Queste unità muscolo-nervo in miniatura riproducevano bassi livelli di acido sialico, difetti di riciclo e iperattività di mTORC1. Trattate con copanlisib durante lo stress da carenza di nutrienti, gli organoidi malati formarono molte più punte di riciclo all’interno delle fibre muscolari, indicando una ripresa della manutenzione cellulare.
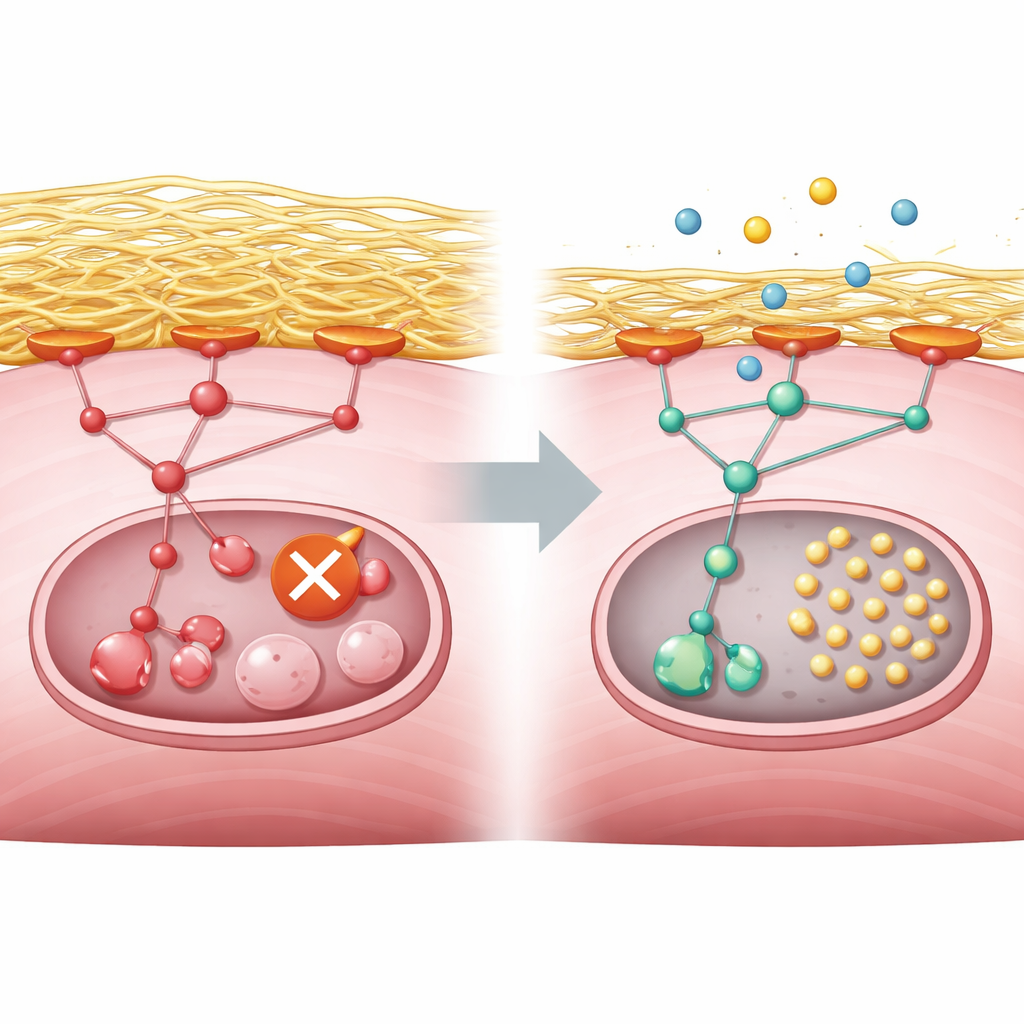
Cosa significa per le persone con miopatia da GNE
In termini semplici, questo lavoro collega un gene difettoso nella sintesi degli zuccheri nelle cellule muscolari a un accumulo eccessivo della matrice circostante, che sovrastimola un percorso di controllo della crescita e spegne la squadra addetta alla pulizia cellulare. Lo studio dimostra che questo stesso schema appare in cellule murine ingegnerizzate, in muscolo derivato da cellule staminali umane, in organoidi e nell’attività genica delle biopsie di pazienti. Sebbene il copanlisib non sia ancora una terapia per la miopatia da GNE, gli esperimenti suggeriscono che una attenuazione mirata della via PI3K–AKT–mTORC1 potrebbe alleviare il blocco del riciclo e potenzialmente rallentare il danno muscolare. Studi clinici futuri dovranno valutare se questo approccio è sicuro ed efficace nelle persone, ma il lavoro fornisce un bersaglio chiaro e verificabile per terapie future.
Citazione: Kim, DW., Kwon, EJ., Kwon, H. et al. Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models. Exp Mol Med 58, 1187–1202 (2026). https://doi.org/10.1038/s12276-026-01701-7
Parole chiave: miopatia da GNE, autofagia muscolare, matrice extracellulare, via AKT mTORC1, copanlisib