Clear Sky Science · fr
Une autophagie défaillante dans la myopathie GNE est restaurée par l’inhibition de l’activation non canonique Akt–mTORC1 dans plusieurs modèles isogéniques
Pourquoi le ménage cellulaire des muscles est important
Nos muscles détruisent en permanence les éléments usés et les reconstruisent. Cet entretien interne, appelé recyclage cellulaire, maintient la solidité des fibres musculaires tout au long de la vie. Dans une affection héréditaire rare connue sous le nom de myopathie GNE, des adultes jeunes perdent progressivement de la force musculaire, mais la chaîne d’événements reliant un seul gène défectueux à l’atrophie n’était pas claire. Cette étude suit cette chaîne pas à pas, en utilisant des modèles issus de cellules souches, des cellules musculaires de souris modifiées et des organoïdes neuromusculaires miniatures, pour montrer comment un environnement perturbé autour de la cellule musculaire peut couper le recyclage à l’intérieur de la cellule et comment un médicament anticancéreux existant pourrait aider à le réactiver.
Du changement génétique à la faiblesse musculaire
La myopathie GNE est causée par des altérations d’un gène qui aide normalement les cellules à fabriquer de l’acide sialique, un petit sucre qui orne de nombreuses molécules de surface. Lorsque le gène fait défaut, les fibres musculaires manquent d’acide sialique et finissent par développer des vacuoles périphériques, des structures en forme de bulles observées au microscope dans les biopsies des patients. Des travaux antérieurs laissaient entendre qu’une autophagie défectueuse pouvait contribuer au problème, mais le câblage précis reliant le défaut génétique à l’échec du recyclage restait inconnu. Les auteurs ont commencé par comparer l’activité génique de cellules musculaires dérivées de cellules souches humaines qui ne différaient que par la présence d’une mutation courante de GNE. Ils ont observé de larges modifications des ensembles de gènes liés au recyclage, suggérant que les voies d’entretien étaient systématiquement perturbées dans les cellules malades.
Construire des modèles cellulaires correspondants
Pour approfondir les mécanismes, l’équipe a créé un système cellulaire musculaire plus simple en supprimant le gène Gne dans une lignée standard de myoblastes de souris. Ces cellules modifiées présentaient la même carence en acide sialique observée dans les modèles humains et chez les patients. En conditions de croissance habituelles, elles paraissaient assez normales, mais lorsque les apports en sucres étaient retirés pour imiter un stress métabolique, les cellules knockout mouraient beaucoup plus facilement. Des mesures protéiques détaillées et des images ont révélé que, dans ces cellules stressées, les structures de recyclage appelées autophagosomes se formaient mal et que le flux normal de matériel vers les compartiments de dégradation cellulaire était ralenti. Cela a confirmé que la défaillance du recyclage est une faiblesse centrale révélée lorsque le muscle déficient en GNE est sollicité par un besoin énergétique.
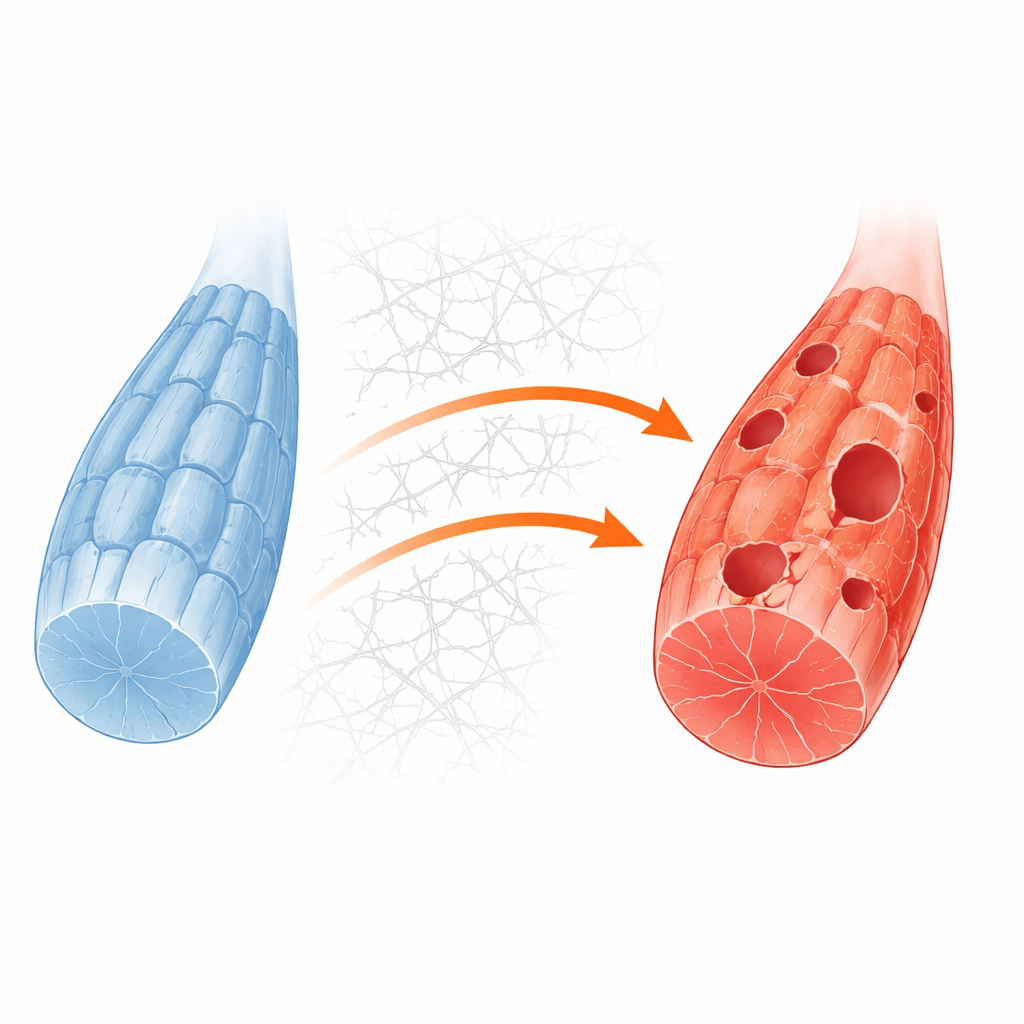
Comment l’extérieur de la cellule grippe l’intérieur
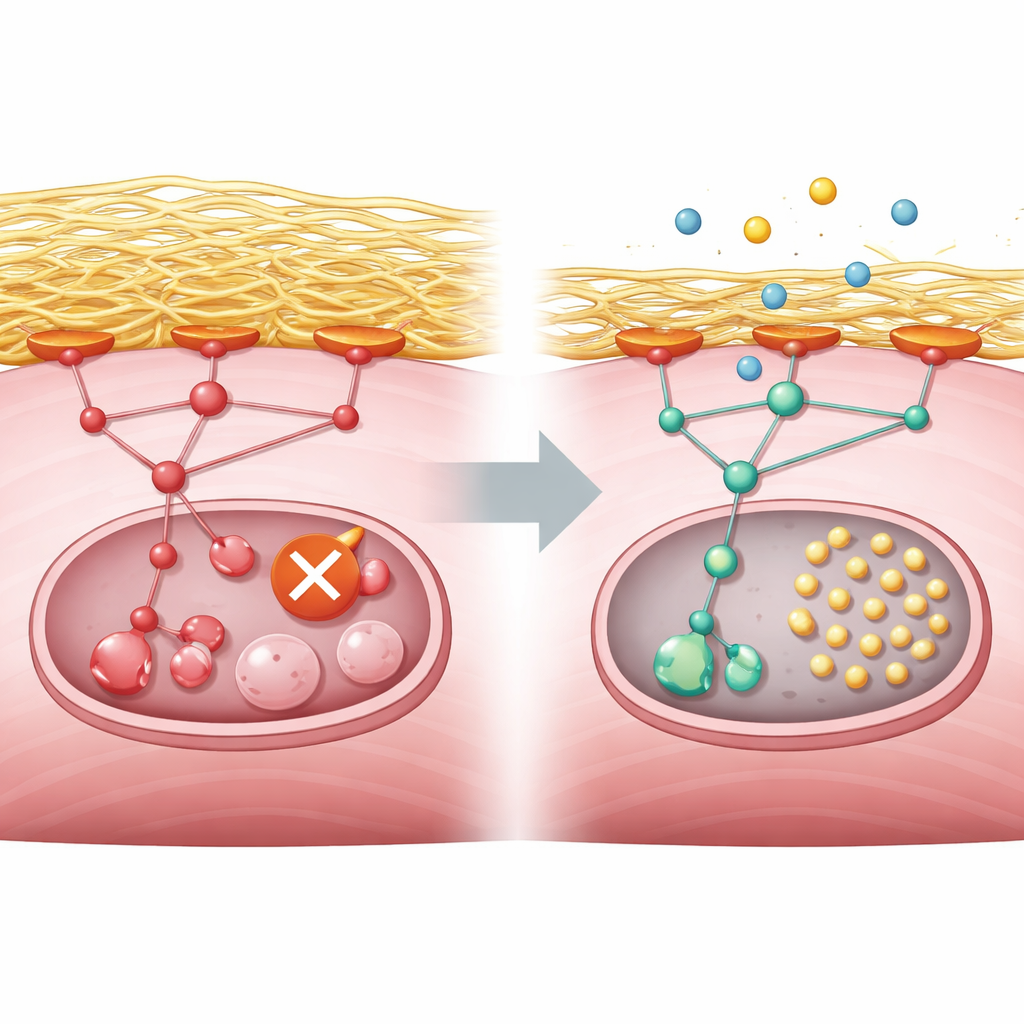
Les chercheurs ont ensuite cherché ce qui bloquait l’autophagie au niveau moléculaire. Leurs analyses ont pointé vers une voie de contrôle de la croissance suractivée, conduite par des protéines connues sous les noms PI3K, AKT et mTORC1, qui répondent normalement aux nutriments et aux signaux de croissance. Dans un muscle sain, la pénurie d’énergie calme cette voie et permet à une enzyme initiatrice appelée ULK1 de déclencher le recyclage. Dans les cellules déficientes en GNE, cependant, la voie restait activée même pendant la privation de nutriments. Parallèlement, les cellules produisaient en excès du collagène et d’autres composants de l’armature environnante, la matrice extracellulaire. Cet excès renforçait les points de contact entre la surface cellulaire et son environnement, ce qui à son tour activait une molécule relais appelée kinases d’adhésion focale (focal adhesion kinase) et alimentait la voie PI3K–AKT–mTORC1. Le résultat était une marque chimique persistante sur ULK1 qui le maintient à l’état inactif, empêchant ainsi le déclenchement du recyclage.
Tester un médicament qui desserre le frein
Munie de ce schéma d’interactions, l’équipe a exploré une large base de données de modifications géniques induites par des médicaments pour trouver des molécules dont les effets s’opposaient au profil de la maladie. Parmi les principaux candidats figuraient de nombreux inhibiteurs de PI3K ou de mTOR, et ils ont sélectionné le copanlisib, un inhibiteur intraveineux de PI3K approuvé pour certains lymphomes, pour des tests supplémentaires. Dans les cellules musculaires de souris knockout GNE, le copanlisib a réduit le signal de voie suractivé, supprimé la marque inhibitrice sur ULK1 et restauré l’activité de recyclage, bien qu’il n’ait pas corrigé la carence initiale en acide sialique. Les auteurs se sont ensuite tournés vers des organoïdes neuromusculaires tridimensionnels issus de cellules souches humaines portant la mutation GNE. Ces mini unités muscle-nerf reproduisaient les faibles niveaux d’acide sialique, les défauts de recyclage et l’hyperactivité de mTORC1. Lorsqu’ils ont été traités par copanlisib pendant un stress nutritif, les organoïdes malades ont formé beaucoup plus de points de recyclage à l’intérieur des fibres musculaires, signe d’un entretien relancé.
Ce que cela signifie pour les personnes atteintes de myopathie GNE
En résumé, ce travail relie un gène de biosynthèse des sucres défectueux dans les cellules musculaires à une accumulation excessive de matrice environnante, qui surexcite une voie de contrôle de la croissance et met hors service l’équipe de nettoyage cellulaire. L’étude montre que ce même schéma apparaît dans des cellules de souris modifiées, des muscles dérivés de cellules souches humaines, des organoïdes et dans l’activité génique provenant de biopsies de patients. Si le copanlisib n’est pas encore un traitement de la myopathie GNE, les expériences suggèrent qu’une atténuation soigneuse de la voie PI3K–AKT–mTORC1 pourrait lever le blocage du recyclage et potentiellement ralentir les dommages musculaires. Des essais cliniques futurs devront déterminer si cette approche est sûre et efficace chez l’humain, mais le travail fournit une cible thérapeutique claire et testable.
Citation: Kim, DW., Kwon, EJ., Kwon, H. et al. Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models. Exp Mol Med 58, 1187–1202 (2026). https://doi.org/10.1038/s12276-026-01701-7
Mots-clés: myopathie GNE, autophagie musculaire, matrice extracellulaire, voie AKT–mTORC1, copanlisib