Clear Sky Science · es
La autofagia defectuosa en la miopatía GNE se restablece al inhibir la activación no canónica Akt–mTORC1 en múltiples modelos isogénicos
Por qué importa la limpieza celular en el músculo
Nuestros músculos descomponen continuamente las piezas desgastadas y las reconstruyen. Esta tarea de mantenimiento interno, llamada reciclaje celular, mantiene las fibras musculares fuertes a lo largo de la vida. En una enfermedad hereditaria rara conocida como miopatía GNE, adultos jóvenes pierden fuerza muscular de forma progresiva, pero la cadena de eventos que va de un único gen defectuoso al desgaste muscular había sido poco clara. Este estudio sigue esa cadena paso a paso, usando modelos de células madre, células musculares de ratón modificadas y organoides neuromusculares en miniatura para mostrar cómo un entorno alterado alrededor de la célula muscular puede apagar el reciclaje dentro de la célula y cómo un fármaco ya utilizado en cáncer podría ayudar a volver a encenderlo.
Del cambio genético a la debilidad muscular
La miopatía GNE es causada por variantes en un gen que normalmente ayuda a las células a sintetizar ácido siálico, un azúcar pequeño que adorna muchas moléculas de la superficie. Cuando el gen falla, las fibras musculares carecen de ácido siálico y con el tiempo desarrollan vacuolas en el borde, estructuras similares a burbujas que se observan al microscopio en biopsias de pacientes. Trabajos previos insinuaban que la autofagia defectuosa podría ser parte del problema, pero el cableado preciso desde el defecto genético hasta la falla del reciclaje era desconocido. Los autores empezaron comparando la actividad génica en células musculares derivadas de células madre humanas que solo diferían en si portaban o no una mutación común de GNE. Observaron amplios cambios en conjuntos de genes relacionados con el reciclaje, lo que sugiere que las vías de mantenimiento estaban sistemáticamente perturbadas en las células enfermas.
Construcción de modelos celulares equivalentes
Para profundizar en los mecanismos, el equipo creó un sistema celular muscular más simple eliminando el gen Gne en una línea estándar de mioblastos de ratón. Estas células modificadas mostraron la misma falta de ácido siálico observada en los modelos humanos y en pacientes. En condiciones de cultivo rutinarias parecían bastante normales, pero cuando se retiraron los suministros de azúcar para imitar el estrés metabólico, las células con el gen eliminado tenían mucha más probabilidad de morir. Mediciones detalladas de proteínas e imágenes revelaron que, en estas células estresadas, las estructuras de reciclaje llamadas autofagosomas se formaban con dificultad y el flujo normal de material hacia los compartimentos de degradación celular estaba reducido. Esto confirmó que la falla del reciclaje es una debilidad central que se hace evidente cuando el músculo deficiente en GNE se ve sometido a demanda energética.
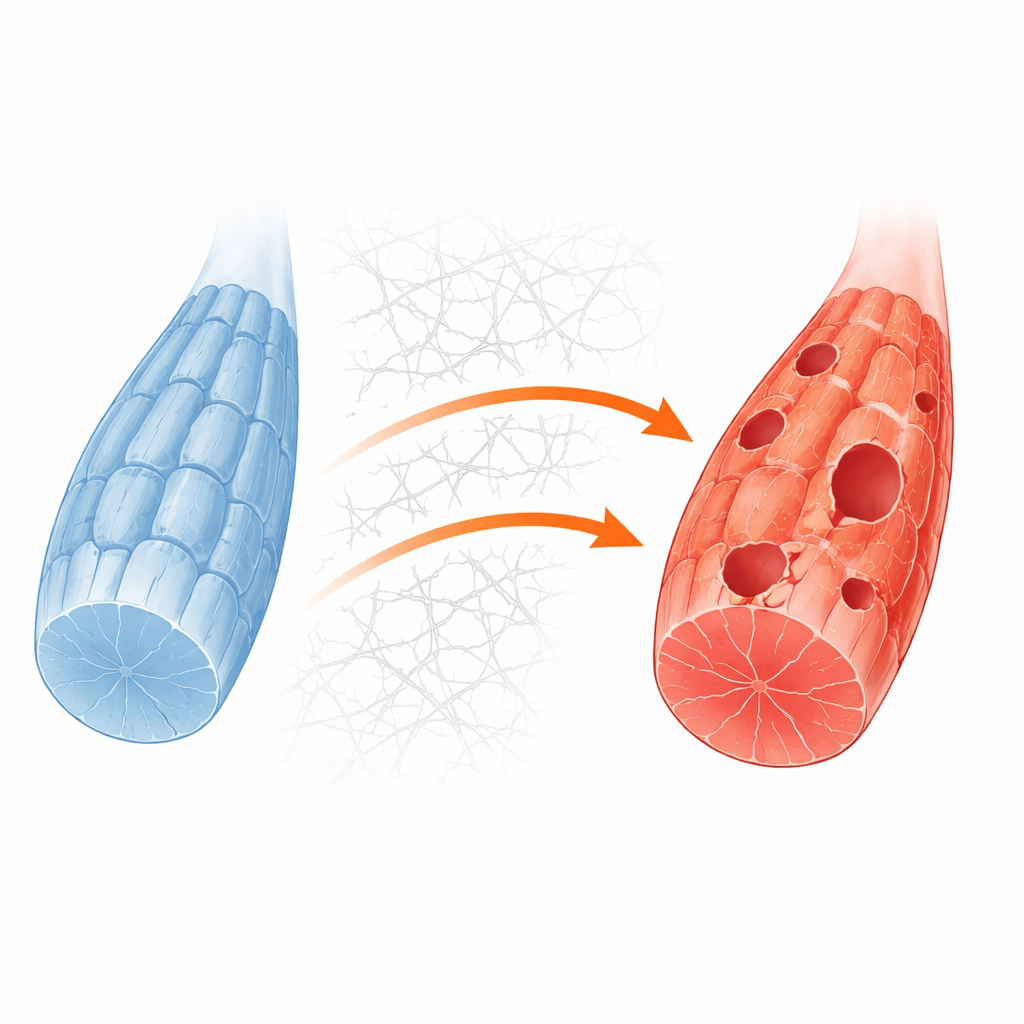
Cómo el exterior de la célula bloquea el interior
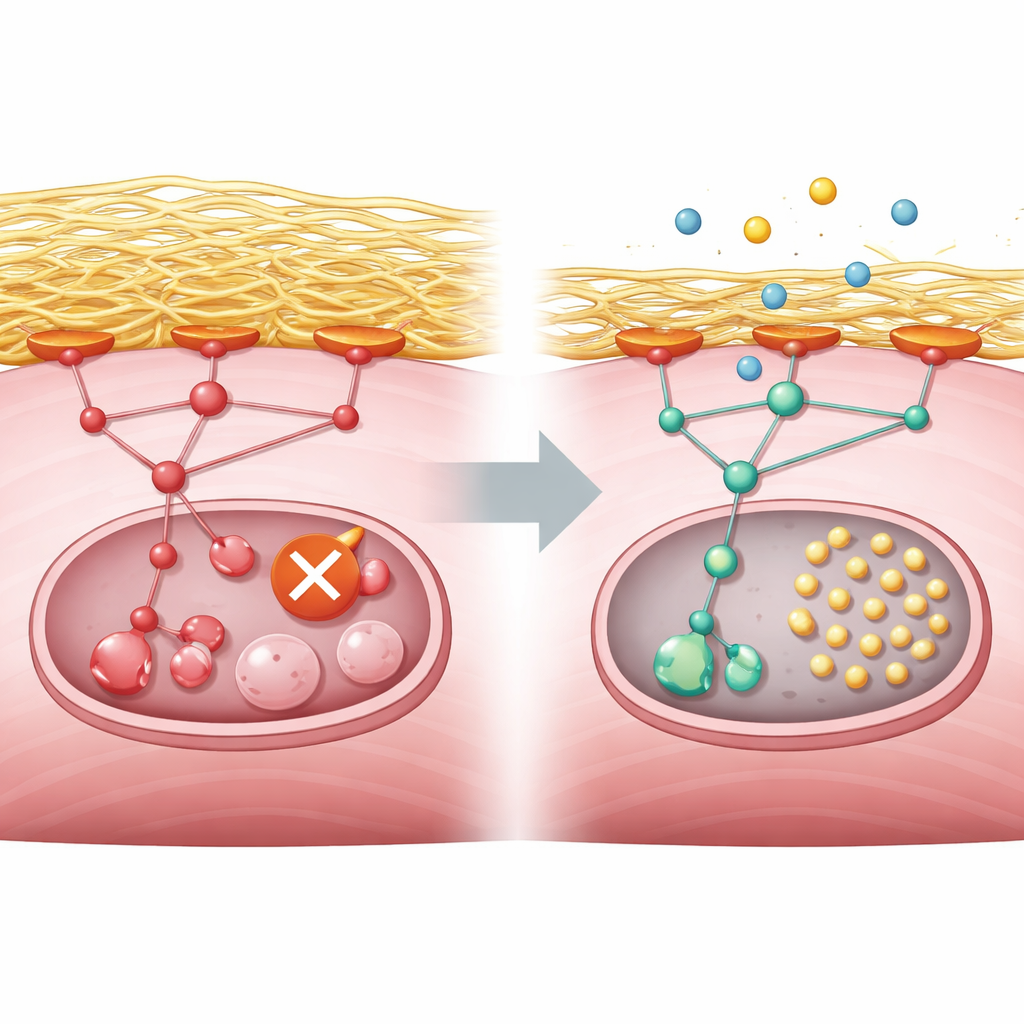
Los investigadores preguntaron a continuación qué estaba bloqueando la autofagia a nivel molecular. Sus análisis señalaron a una vía de control del crecimiento sobreactivada impulsada por proteínas conocidas como PI3K, AKT y mTORC1, que normalmente responden a nutrientes y señales de crecimiento. En músculo sano, la falta de energía silencia esta vía y permite que una enzima iniciadora llamada ULK1 active el reciclaje. Sin embargo, en las células deficientes en GNE la vía permanecía encendida incluso durante el ayuno. Al mismo tiempo, las células sobreproducían colágeno y otros componentes del andamiaje circundante, la matriz extracelular. Este exceso de andamiaje reforzó los puntos de contacto entre la superficie celular y su entorno, lo que a su vez activó una molécula de retransmisión llamada quinasa de adhesión focal y alimentó la vía PI3K–AKT–mTORC1. El resultado fue una marca química persistente en ULK1 que lo mantiene en estado inactivo, impidiendo que el reciclaje siquiera comience.
Prueba de un fármaco que afloja el freno
Con este diagrama de conexiones, el equipo buscó en una gran base de datos de cambios génicos inducidos por fármacos para encontrar medicinas cuyos efectos fueran opuestos a la firma de la enfermedad. Muchos de los principales candidatos eran inhibidores de PI3K o mTOR, y seleccionaron copanlisib, un inhibidor intravenoso de PI3K aprobado para ciertos linfomas, para pruebas adicionales. En células musculares de ratón con eliminación de Gne, copanlisib redujo la señal sobreactivada de la vía, eliminó la marca inhibitoria de ULK1 y restauró la actividad de reciclaje, aunque no reparó la deficiencia original de ácido siálico. Los autores pasaron luego a organoides neuromusculares tridimensionales derivados de células madre humanas portadoras de la mutación GNE. Estas unidades músculo-nervio en miniatura reprodujeron los bajos niveles de ácido siálico, los defectos de reciclaje y la sobreactividad de mTORC1. Al tratar los organoides enfermos con copanlisib durante el estrés nutritivo, formaron muchas más puntas de reciclaje dentro de las fibras musculares, lo que indica una recuperación del mantenimiento celular.
Qué significa esto para las personas con miopatía GNE
En términos sencillos, este trabajo vincula un gen defectuoso implicado en la síntesis de un azúcar en las células musculares con una acumulación excesiva del andamiaje circundante, que sobreestimula una vía de control del crecimiento y apaga el equipo de limpieza de la célula. El estudio muestra que este mismo patrón aparece en células de ratón modificadas, en músculo derivado de células madre humanas y en organoides, así como en la actividad génica de biopsias de pacientes. Aunque copanlisib aún no es un tratamiento para la miopatía GNE, los experimentos sugieren que atenuar con cuidado la vía PI3K–AKT–mTORC1 podría aliviar el bloqueo del reciclaje y potencialmente frenar el daño muscular. Estudios clínicos futuros deberán determinar si este enfoque es seguro y eficaz en personas, pero el trabajo proporciona un blanco claro y comprobable para la terapia.
Cita: Kim, DW., Kwon, EJ., Kwon, H. et al. Defective autophagy in GNE myopathy is rescued by inhibition of noncanonical Akt–mTORC1 activation across multiple isogenic models. Exp Mol Med 58, 1187–1202 (2026). https://doi.org/10.1038/s12276-026-01701-7
Palabras clave: Miopatía GNE, autofagia muscular, matriz extracelular, vía AKT–mTORC1, copanlisib