Clear Sky Science · sv
En kvantmekanisk ram för biologiska system i skala med miljoner atomer
Att skåda in i livet, en elektron i taget
Proteiner, DNA och virus styrs alla av elektronernas ständiga rörelse, men verkligt kvantmekaniska simuleringar av så stora molekyler har länge varit för långsamma för annat än de minsta systemen. Detta arbete introducerar ett sätt att föra full kvantmekanik till enorma biologiska strukturer — upp till tiotals miljoner atomer — genom att varsamt släppa på precisionen precis så mycket att beräkningarna blir snabba, prisvärda och överraskande användbara för frågor inom biologi, medicin och läkemedelsdesign.
Varför stora molekyler behöver kvantögon
Många biologiska händelser, från läkemedelsbindning till ljusets effekt på synen, innebär att kemiska bindningar bryts och bildas och att elektronmoln förskjuts. Klassiska simuleringsmetoder kan följa atomernas övergripande rörelser i stora proteiner eller DNA, men de behandlar elektronerna indirekt och missar vissa av de finare effekter som bestämmer hur ljus absorberas eller hur tätt ett läkemedel sitter fast vid sitt mål. Kvantmekaniska metoder spårar elektronerna explicit och kan förutsäga egenskaper som optiska spektra med hög trohet. Problemet är att traditionella kvantmetoder blir mycket långsamma när systemen växer, så de har i regel bara använts för små fragment, vilket lämnar hela virus eller stora proteinaggregat utom räckhåll.
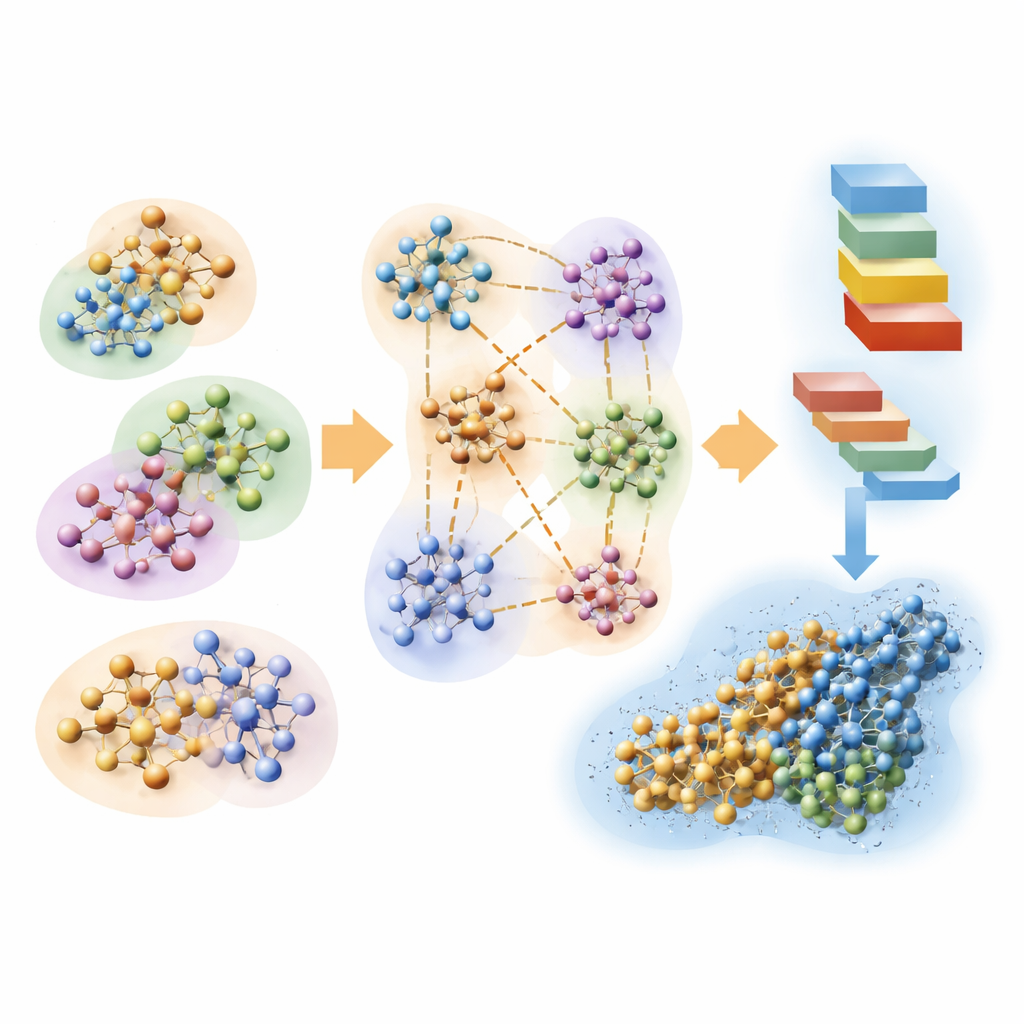
Att dela upp problemet i hanterbara bitar
Författarna bygger vidare på en klassisk kvantmetod kallad Hartree–Fock, som beskriver elektroner via matematiska orbitaler, och formar om den för hastighet snarare än maximal precision. Deras huvudidé är att dela upp en enorm biomolekyl plus dess omgivande vatten i många överlappande kluster. Varje kluster har en central kärnregion där de kvantmekaniska resultaten bevaras, och en buffertregion som efterliknar påverkan från närliggande atomer. Kvantberäkningar utförs sedan separat för varje kluster, och bitarna sys ihop för att rekonstruera hela systemets elektroniska densitet. Genom att även använda en mycket kompakt representation av orbitalerna och skära bort svaga, långräckviddiga interaktioner som bidrar lite till slutresultatet, skalar metoden ungefär i proportion till antalet atomer i stället för att explodera i kostnad.
Från virus till DNA och cancerläkemedel
För att demonstrera vad denna strömlinjeformade ram klarar av tar forskarna sig an flera demonstrationssystem. De beräknar hela den elektroniska strukturen för tre stora sammanställningar i vatten, inklusive en bakteriofag — ett virus som infekterar bakterier — tillsammans med en omgivande lösningslåda som totalt uppgår till mer än 45 miljoner atomer och över 150 miljoner elektroner. På en modern klusterdator slutförs denna beräkning på ungefär en halv dag, vilket enligt deras kännedom är den största Hartree–Fock-simulering som någonsin utförts. De vänder sig sedan till ljusabsorption: med en tidsberoende förlängning av sin metod simulerar de ultraviolett–synliga spektra för DNA-fragment på upp till 21 baspar och för cancerläkemedlet actinomycin D, både fritt och bundet till DNA. De förutsagda spektrumen matchar experimentella mätningar väl, vilket visar att den reducerade-precisionen fortfarande fångar den väsentliga fysiken för hur dessa biomolekyler interagerar med ljus.
Kontrollera AI-förutsagda proteinformer med fysik
Teamet undersöker också hur snabba kvantberäkningar kan hjälpa till att utvärdera proteinstrukturer förutspådda av artificiella intelligensverktyg som AlphaFold. Proteiner veckar sig naturligt till former som minimerar deras energi. Med sin linjärt skalande Hartree–Fock-ansats uppskattar författarna lokala atomenergier längs flera AlphaFold-förutspådda proteiner i realistiska vattenmiljöer. När dessa kvantbaserade energimönster jämförs med AlphaFolds egna förtroendepoäng överensstämmer de slagkraftigt: låga energier sammanfaller med regioner AlphaFold anser vara pålitliga, medan höga energier markerar osäkra eller instabila segment. Detta tyder på att moderna nätverk för strukturprediktion implicit lär sig aspekter av den underliggande kvantenergilandskapet, och att förstaprincipberäkningar kan ge en oberoende, fysikbaserad kvalitetskontroll.
Avvägningar, begränsningar och nya möjligheter
Hastighetsvinsterna kommer från medvetna kompromisser. Att använda en minimal orbitalbas och att kapa bort svaga, avlägsna interaktioner innebär nödvändigtvis en viss förlust i noggrannhet, och del-och-härska-strategin är inte idealisk för storheter som känsligt beror på långräckviddseffekter, såsom precisa bindningsenergier i täta protein–ligand-komplex. För sådana högprecisionsuppgifter visar författarna att avkapningarna måste mildras eller tas bort, och metoden uppträder då mer som en konventionell, långsammare kvantkod. Ändå, för mycket stora system där ingen fullständigt exakt kvantbehandling alls är möjlig, erbjuder deras angreppssätt en kraftfull mellanväg: det levererar kvalitativt och ofta kvantitativt tillförlitliga insikter med måttliga datorresurser, till och med ned till enstaka beräkningsnoder för system upp till ungefär en miljon atomer.
Vad detta innebär för molekylärvetenskapens framtid
Genom att omkonstruera en standardkvantmetod för att prioritera hastighet samtidigt som man håller god överensstämmelse med experiment, öppnar denna ram dörren för rutinmässiga kvantnivåstudier av hela virus, proteinmaskiner och stora DNA–läkemedelskomplex. Den möjliggör detaljerade elektron-densitetskartor som kan jämföras direkt med modern kristallografi, realistiska optiska spektra för biomolekyler i en aldrig tidigare skådad storlek och oberoende kontroller av AI-genererade proteinstrukturer. Framöver kan samma idéer stödja snabba kvantbaserade molekylärdynamikstudier, undersökningar av hur elektriska eller magnetiska fält påverkar biomolekyler och generering av stora, kvantkorrekta datamängder för att träna nästa generations maskininlärningsmodeller. I praktiska termer förflyttar det den tidigare avlägsna drömmen om ”kvantbiologi” — att behandla livets maskineri med full elektronisk detalj — närmare vardaglig vetenskaplig praxis.
Citering: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Nyckelord: kvant biomolekylär simulering, Hartree–Fock, prediktion av proteinstruktur, UV-Vis-spektra, del-och-härska-algoritmer