Clear Sky Science · pt
Um arcabouço quântico-mecânico para sistemas biológicos em escala de milhões de átomos
Olhar para dentro da vida, um elétron de cada vez
Proteínas, DNA e vírus devem seu comportamento ao movimento incessante dos elétrons, mas simulações verdadeiramente em nível quântico de moléculas tão grandes há muito tempo são lentas demais para rodar em quaisquer sistemas que não os menores. Este trabalho apresenta uma maneira de levar a mecânica quântica completa a estruturas biológicas enormes — de até dezenas de milhões de átomos — relaxando cuidadosamente a precisão o suficiente para tornar os cálculos rápidos, acessíveis e surpreendentemente úteis para questões em biologia, medicina e desenho de fármacos.
Por que grandes moléculas precisam de olhos quânticos
Muitos eventos biológicos, desde a ligação de um fármaco até a ação da luz na visão, envolvem quebrar e formar ligações químicas e deslocar nuvens de elétrons. Métodos clássicos de simulação conseguem acompanhar o movimento geral dos átomos em grandes proteínas ou no DNA, mas tratam os elétrons apenas indiretamente e deixam passar alguns dos efeitos mais sutis que determinam como a luz é absorvida ou quão firmemente um fármaco se prende ao seu alvo. Em contraste, abordagens quântico-mecânicas rastreiam elétrons explicitamente e podem prever propriedades como espectros ópticos com alta fidelidade. O problema é que métodos quânticos tradicionais tornam-se extremamente lentos conforme os sistemas crescem, de modo que tipicamente foram usados só em fragmentos pequenos, deixando vírus inteiros ou enormes agregados de proteínas fora de alcance.
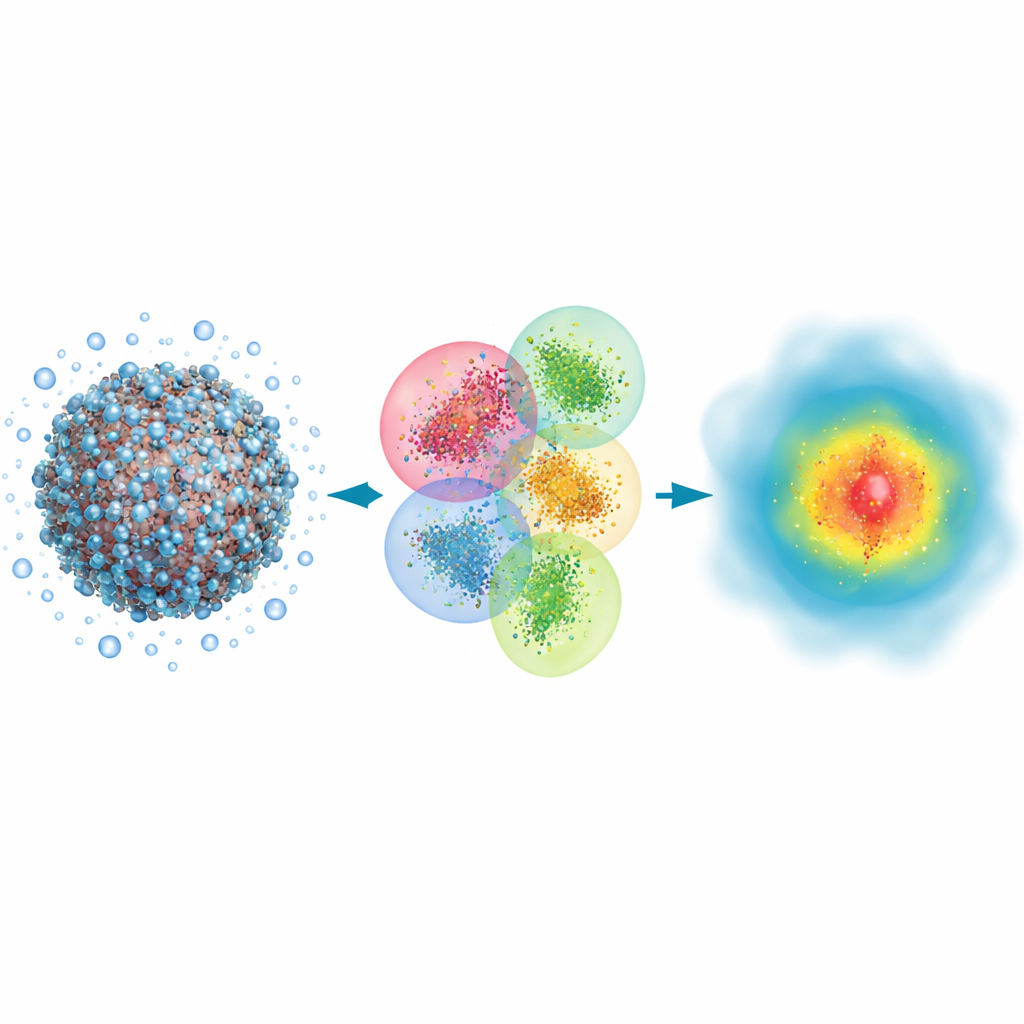
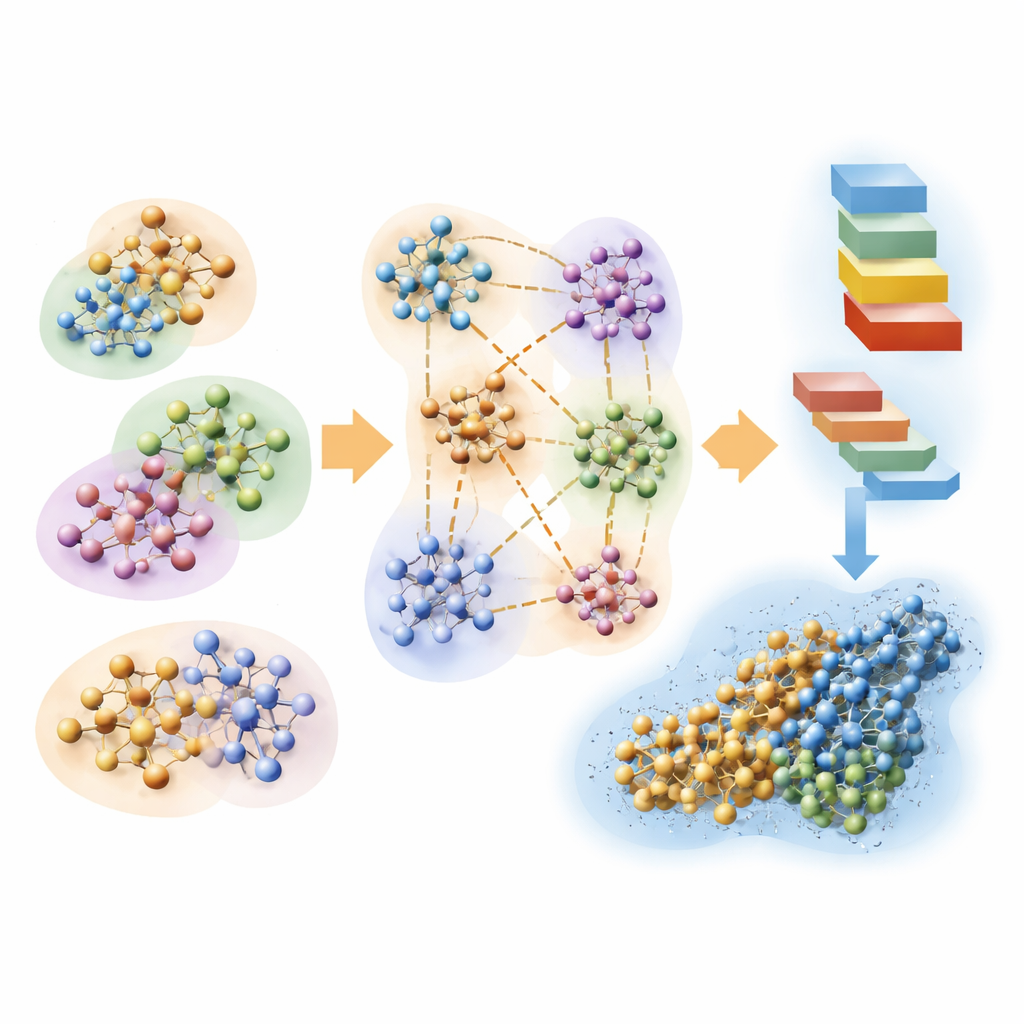
Quebrando o problema em pedaços manejáveis
Os autores partem de uma abordagem quântica clássica chamada Hartree–Fock, que descreve elétrons por orbitais matemáticos, e a reformulam para velocidade em vez de precisão máxima. A ideia-chave é dividir uma biomolécula enorme mais a água ao redor em muitos aglomerados sobrepostos. Cada aglomerado tem uma região central onde os resultados quânticos são mantidos e uma região de amortecimento que imita a influência dos átomos vizinhos. Cálculos quânticos são então realizados separadamente em cada aglomerado, e as peças são costuradas para reconstruir a densidade eletrônica completa do sistema. Ao usar também uma representação muito compacta dos orbitais e descartar interações fracas e de longo alcance que contribuem pouco para a resposta final, o método escala aproximadamente em proporção ao número de átomos em vez de explodir em custo.
De vírus a DNA e medicamentos contra o câncer
Para demonstrar o que esse arcabouço simplificado pode fazer, os pesquisadores atacam vários sistemas exemplares. Eles calculam a estrutura eletrônica completa de três grandes agregados em água, incluindo um bacteriófago — um vírus que infecta bactérias — juntamente com uma caixa de solvente ao redor totalizando mais de 45 milhões de átomos e mais de 150 milhões de elétrons. Em um cluster moderno esse cálculo termina em cerca de meio dia, representando, segundo seus relatos, a maior simulação Hartree–Fock já realizada. Em seguida tratam da absorção de luz: usando uma extensão dependente do tempo de seu método, simulam espectros ultravioleta–visíveis para fragmentos de DNA de até 21 pares de bases e para o fármaco anticâncer Actinomicina D, tanto livre quanto ligado ao DNA. Os espectros previstos se concordam estreitamente com medidas experimentais, mostrando que o esquema de precisão reduzida ainda captura a física essencial de como essas biomoléculas interagem com a luz.
Conferindo formas de proteínas previstas por IA com a física
A equipe também explora como cálculos quânticos rápidos podem ajudar a avaliar estruturas de proteínas previstas por ferramentas de inteligência artificial como o AlphaFold. Proteínas naturalmente se dobram em formas que minimizam sua energia. Usando sua abordagem Hartree–Fock de escalonamento linear, os autores estimam energias atômicas locais ao longo de várias proteínas previstas pelo AlphaFold, em ambientes aquosos realistas. Quando esses padrões de energia derivados da mecânica quântica são comparados com as próprias pontuações de confiança do AlphaFold, eles se alinham de maneira marcante: energias baixas coincidem com regiões que o AlphaFold considera confiáveis, enquanto energias altas sinalizam segmentos incertos ou instáveis. Isso sugere que as redes modernas de predição estrutural estão aprendendo implicitamente aspectos da paisagem de energia quântica subjacente, e que cálculos a partir de princípios fundamentais podem fornecer uma verificação independente e baseada na física.
Compromissos, limites e novas possibilidades
Os ganhos de velocidade vêm de concessões deliberadas. Usar uma base de orbitais mínima e cortar interações fracas e distantes sacrifica necessariamente alguma precisão, e a estratégia dividir-e-conquistar não é ideal para quantidades que dependem delicadamente de efeitos de longo alcance, como energias de ligação precisas em complexos proteína–ligante muito apertados. Os autores mostram que, para tais tarefas de alta precisão, os cortes precisam ser relaxados ou removidos, e o método passa a se comportar mais como um código quântico convencional e mais lento. Ainda assim, para sistemas muito grandes em que nenhum tratamento quântico totalmente preciso é viável, a abordagem oferece um meio-termo poderoso: fornece insights qualitativos e frequentemente quantitativos confiáveis com recursos computacionais modestos, mesmo em nós de computação únicos para sistemas de até aproximadamente um milhão de átomos.
O que isso significa para o futuro da ciência molecular
Ao reengenheirar um método quântico padrão para favorecer velocidade mantendo bom acordo com experimentos, esse arcabouço abre a porta para estudos rotineiros em nível quântico de vírus inteiros, máquinas proteicas e grandes complexos DNA–fármaco. Ele possibilita mapas detalhados de densidade eletrônica que podem ser comparados diretamente com cristalografia de ponta, espectros ópticos realistas para biomoléculas de tamanho sem precedentes e verificações independentes de estruturas de proteínas geradas por IA. Olhando adiante, as mesmas ideias poderiam suportar dinâmicas moleculares rápidas baseadas em quântica, estudos de como campos elétricos ou magnéticos influenciam biomoléculas e a geração de grandes conjuntos de dados com precisão quântica para treinar modelos de aprendizado de máquina de próxima geração. Na prática, aproxima o sonho antes distante da "biologia quântica" — tratar a maquinaria da vida com detalhe eletrônico completo — da prática científica cotidiana.
Citação: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Palavras-chave: simulação biomolecular quântica, Hartree–Fock, predição de estruturas de proteínas, espectros UV-Vis, algoritmos dividir-e-conquistar