Clear Sky Science · nl
Een kwantummechanisch raamwerk voor biologische systemen op miljoenen-atomen schaal
Leven van binnen bekijken, één elektron per keer
Eiwitten, DNA en virussen danken hun gedrag allemaal aan de rusteloze beweging van elektronen, maar echte kwantumniveau-simulaties van zulke grote moleculen waren lange tijd te traag om op andere systemen dan de allerkleinsten te draaien. Dit werk introduceert een manier om volledige kwantummechanica toe te passen op enorme biologische structuren—tot tientallen miljoenen atomen—door de nauwkeurigheid zorgvuldig net genoeg te versoepelen om de berekeningen snel, betaalbaar en verrassend nuttig te maken voor vragen in biologie, geneeskunde en geneesmiddelenontwerp.
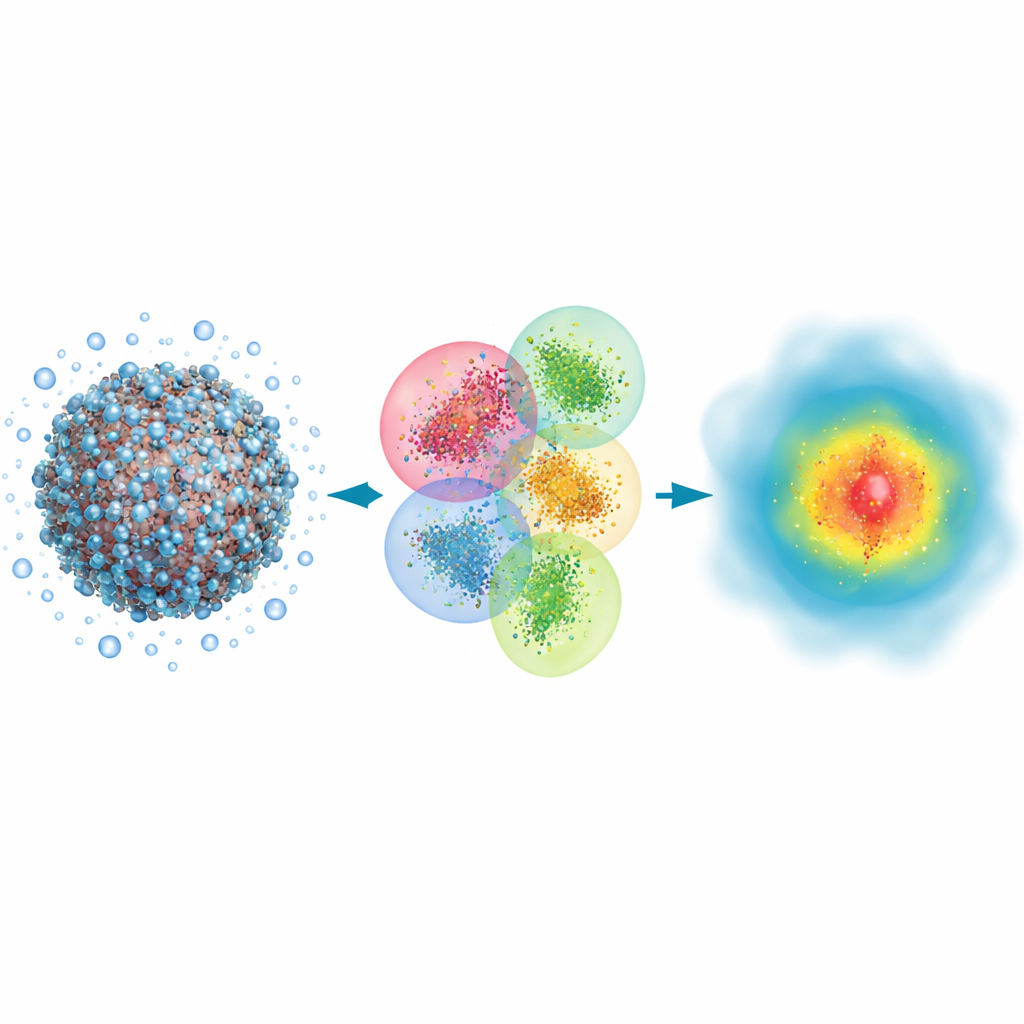
Waarom grote moleculen kwantumbewust moeten worden bekeken
Veel biologische processen, van geneesmiddelbinding tot de werking van zonlicht bij het zien, omvatten het breken en vormen van chemische bindingen en het verschuiven van elektronenwolken. Klassieke simulatietechnieken kunnen de algemene beweging van atomen in grote eiwitten of DNA volgen, maar ze behandelen elektronen slechts indirect en missen sommige subtielere effecten die bepalen hoe licht wordt geabsorbeerd of hoe sterk een geneesmiddel aan zijn doel bindt. Daarentegen volgen kwantummechanische benaderingen elektronen expliciet en kunnen ze eigenschappen zoals optische spectra nauwkeurig voorspellen. Het probleem is dat traditionele kwantummethoden dramatisch trager worden naarmate systemen groeien, zodat ze doorgaans alleen voor kleine fragmenten zijn gebruikt en volledige virussen of enorme eiwitcomplexen buiten bereik bleven.
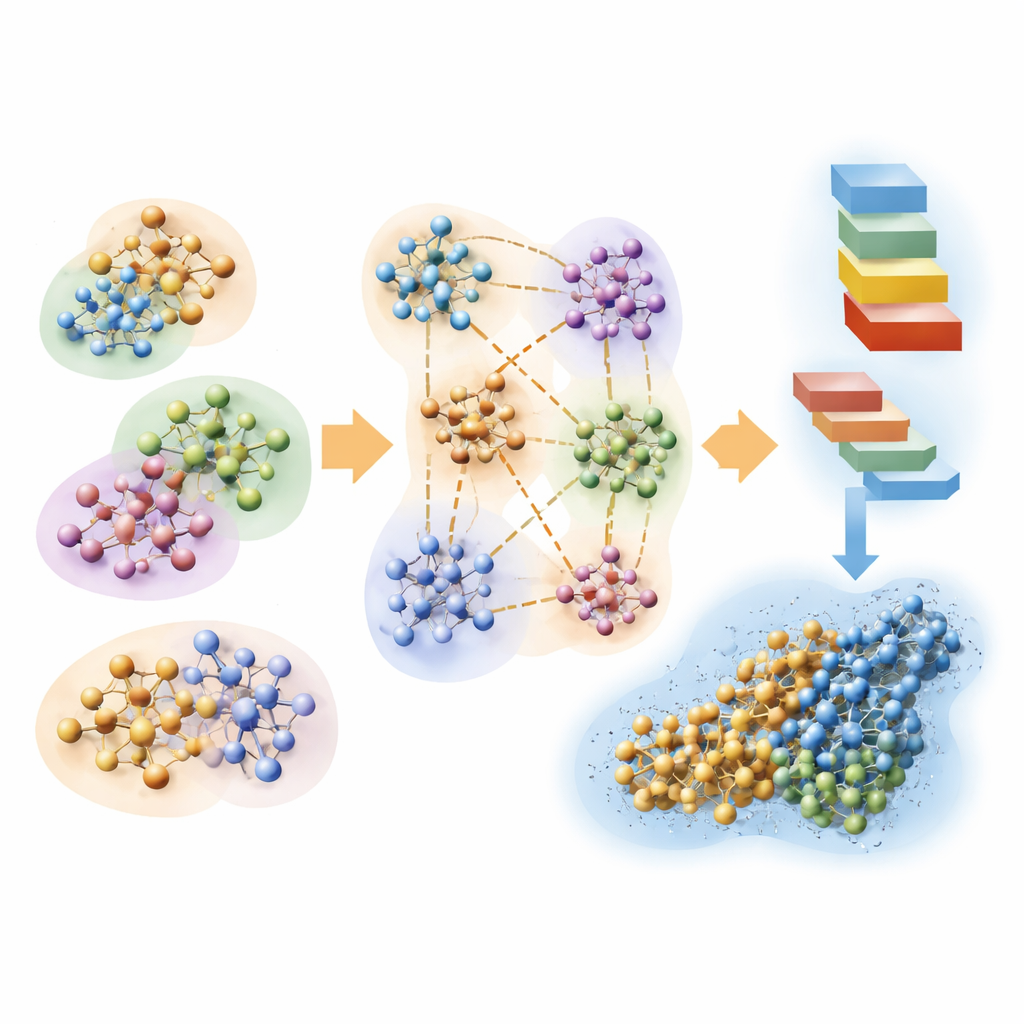
Het probleem in hapklare stukjes breken
De auteurs bouwen voort op een klassieke kwantumaanpak genaamd Hartree–Fock, die elektronen beschrijft met wiskundige orbitalen, en vormen die aanpak om met het oog op snelheid in plaats van ultieme precisie. Hun kernidee is een enorm biomolecuul plus omringend water op te delen in veel overlappende clusters. Elk cluster heeft een centraal kerngebied waarin de kwantumresultaten behouden blijven, en een bufferzone die de invloed van naburige atomen nabootst. Op elk cluster worden vervolgens afzonderlijke kwantumberekeningen uitgevoerd, en de stukjes worden aan elkaar gehecht om de volledige elektrondichtheid van het systeem te reconstrueren. Door ook een zeer compacte representatie van de orbitalen te gebruiken en zwakke, langafstandseffecten die weinig bijdragen aan het eindresultaat weg te laten, schaalt de methode ruwweg in verhouding tot het aantal atomen in plaats van dat de kosten exploderen.
Van virussen tot DNA en kankerremmers
Om te laten zien wat dit gestroomlijnde raamwerk kan, nemen de onderzoekers verschillende aansprekende systemen onder handen. Ze berekenen de volledige elektronische structuur van drie grote assemblages in water, waaronder een bacteriofaag—een virus dat bacteriën infecteert—samen met een omliggende solventbox met in totaal meer dan 45 miljoen atomen en meer dan 150 miljoen elektronen. Op een modern cluster is deze berekening afgerond in ongeveer een halve dag, wat, voor zover zij weten, de grootste ooit uitgevoerde Hartree–Fock-simulatie vertegenwoordigt. Daarna richten ze zich op lichtabsorptie: met een tijdsafhankelijke uitbreiding van hun methode simuleren ze ultraviolet–vis-spectra voor DNA-fragmenten tot 21 basenparen en voor het anticancermiddel actinomycine D, zowel vrij als gebonden aan DNA. De voorspelde spectra sluiten nauw aan bij experimentele metingen, wat aantoont dat het schema met verminderde nauwkeurigheid nog steeds de essentiële fysica van hoe deze biomoleculen met licht interacteren vastlegt.
AI-voorspelde eiwitvormen fysisch controleren
Het team onderzoekt ook hoe snelle kwantumberekeningen kunnen helpen bij het evalueren van eiwitstructuren die voorspeld zijn door AI-tools zoals AlphaFold. Eiwitten vouwen zich van nature in vormen die hun energie minimaliseren. Met hun lineair-schalende Hartree–Fock-aanpak schatten de auteurs lokale atomaire energieën langs de lengte van verschillende door AlphaFold voorspelde eiwitten, in realistische wateromgevingen. Wanneer deze kwantumafgeleide energiemotieven worden vergeleken met AlphaFold’s eigen betrouwbaarheidscores, komen ze opvallend goed overeen: lage energieën vallen samen met regio’s die AlphaFold als betrouwbaar beschouwt, terwijl hoge energieën onzekere of onstabiele segmenten aangeven. Dit suggereert dat moderne structuurvoorspellende netwerken implliciet aspecten van het onderliggende kwantum-energielandschap leren, en dat eerst-principes berekeningen een onafhankelijke, op natuurkunde gebaseerde kwaliteitscontrole kunnen bieden.
Compromissen, beperkingen en nieuwe mogelijkheden
De snelheidswinst komt door bewuste compromissen. Het gebruik van een minimale orbitaalbasis en het wegknippen van zwakke, verre interacties gaat noodzakelijkerwijs ten koste van enige nauwkeurigheid, en de divide-and-conquer-strategie is niet ideaal voor grootheden die gevoelig afhangen van langafstandseffecten, zoals precieze bindingsenergieën in nauwe eiwit–ligandcomplexen. De auteurs tonen aan dat voor zulke hoogprecisie-taken de afkappunten versoepeld of verwijderd moeten worden, waarna de methode zich meer als een conventionele, tragere kwantumcode gedraagt. Toch biedt hun aanpak voor zeer grote systemen waarvoor geen volledig nauwkeurige kwantumbehandeling haalbaar is een krachtig middenweg: het levert kwalitatief en vaak kwantitatief betrouwbare inzichten met bescheiden rekenmiddelen, zelfs tot enkele compute-nodes voor systemen tot ongeveer een miljoen atomen.
Wat dit betekent voor de toekomst van moleculaire wetenschap
Door een standaard kwantummethode te herontwerpen om snelheid te bevoordelen terwijl goede overeenstemming met experimenten behouden blijft, opent dit raamwerk de deur naar routinematige kwantumniveau-studies van volledige virussen, eiwitmachines en grote DNA–geneesmiddelcomplexen. Het maakt gedetailleerde elektrondichtheidskaarten mogelijk die rechtstreeks vergeleken kunnen worden met state-of-the-art kristallografie, realistische optische spectra voor biomoleculen van ongekende grootte en onafhankelijke controles van door AI gegenereerde eiwitstructuren. Vooruitkijkend zouden dezelfde ideeën snelle kwantumgebaseerde moleculaire dynamica kunnen ondersteunen, studies naar hoe elektrische of magnetische velden biomoleculen beïnvloeden, en het genereren van grote, kwantumnauwkeurige datasets om de volgende generatie machine-learningmodellen te trainen. In praktische zin brengt het de ooit verafgelegen droom van “kwantumbiologie”—het behandelen van het levensmechanisme met volledige elektronische detail—dichter bij alledaagse wetenschappelijke praktijk.
Bronvermelding: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Trefwoorden: kwantum bio-moleculaire simulatie, Hartree–Fock, eiwitstructuurvoorspelling, UV-Vis spectra, divide-and-conquer-algoritmen