Clear Sky Science · en
A quantum-mechanical framework for million-atom scale biological systems
Peering Inside Life, One Electron at a Time
Proteins, DNA and viruses all owe their behavior to the restless motion of electrons, but truly quantum-level simulations of such big molecules have long been too slow to run on anything but the smallest systems. This work introduces a way to bring full quantum mechanics to enormous biological structures—up to tens of millions of atoms—by carefully relaxing accuracy just enough to make the calculations fast, affordable and surprisingly useful for questions in biology, medicine and drug design.
Why Big Molecules Need Quantum Eyes
Many biological events, from drug binding to the action of sunlight on vision, involve breaking and making chemical bonds and shifting clouds of electrons. Classical simulation methods can follow the overall motion of atoms in large proteins or DNA, but they treat electrons only indirectly and miss some of the subtler effects that determine how light is absorbed or how tightly a drug clings to its target. In contrast, quantum-mechanical approaches track electrons explicitly and can predict properties such as optical spectra with high fidelity. The catch is that traditional quantum methods slow to a crawl as systems grow, so they have typically been used only for small fragments, leaving entire viruses or huge protein assemblies out of reach.
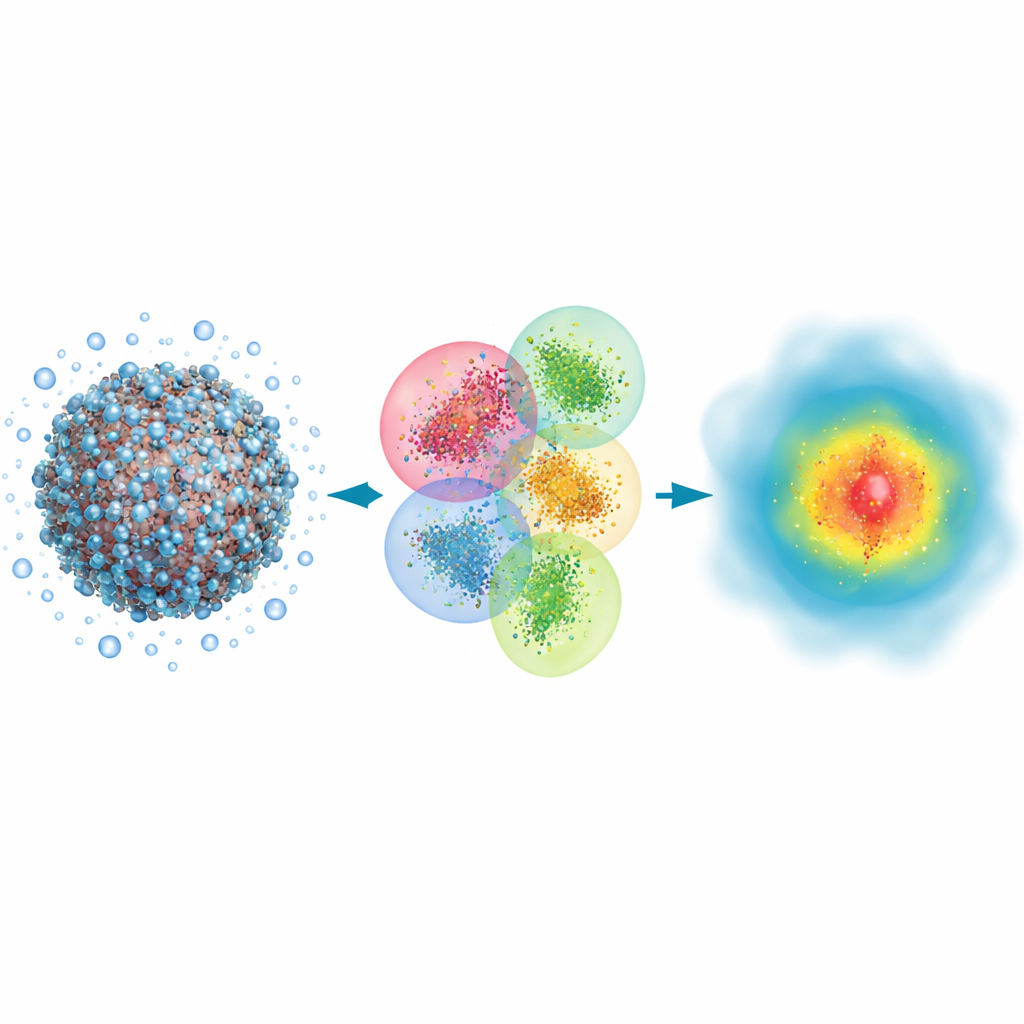
Breaking the Problem into Bite-Sized Pieces
The authors build on a classic quantum approach called Hartree–Fock, which describes electrons using mathematical orbitals, and reshape it for speed rather than ultimate precision. Their key idea is to split a huge biomolecule plus its surrounding water into many overlapping clusters. Each cluster has a central core region where the quantum results are kept, and a buffer region that mimics the influence of neighboring atoms. Quantum calculations are then carried out separately on each cluster, and the pieces are stitched together to reconstruct the full electron density of the system. By also using a very compact representation of the orbitals and discarding weak, long-range interactions that contribute little to the final answer, the method scales roughly in proportion to the number of atoms instead of exploding in cost.
From Viruses to DNA and Cancer Drugs
To demonstrate what this streamlined framework can do, the researchers tackle several showcase systems. They compute the full electronic structure of three large assemblies in water, including a bacteriophage—a virus that infects bacteria—together with a surrounding box of solvent totalling more than 45 million atoms and over 150 million electrons. On a modern cluster this calculation finishes in about half a day, representing, to their knowledge, the largest Hartree–Fock simulation ever performed. They then turn to light absorption: using a time-dependent extension of their method, they simulate ultraviolet–visible spectra for DNA fragments of up to 21 base pairs and for the anticancer drug Actinomycin D, both free and bound to DNA. The predicted spectra closely match experimental measurements, showing that the reduced-accuracy scheme still captures the essential physics of how these biomolecules interact with light.
Checking AI-Predicted Protein Shapes with Physics
The team also explores how fast quantum calculations can help evaluate protein structures predicted by artificial intelligence tools such as AlphaFold. Proteins naturally fold into shapes that minimize their energy. Using their linear-scaling Hartree–Fock approach, the authors estimate local atomic energies along the length of several AlphaFold-predicted proteins, in realistic water environments. When these quantum-derived energy patterns are compared with AlphaFold’s own confidence scores, they align strikingly well: low energies coincide with regions AlphaFold deems reliable, while high energies flag uncertain or unstable segments. This suggests that modern structure-prediction networks are implicitly learning aspects of the underlying quantum energy landscape, and that first-principles calculations can provide an independent, physics-based quality check.
Trade-Offs, Limits and New Possibilities
The speed gains come from conscious compromises. Using a minimal orbital basis and cutting off weak, distant interactions necessarily sacrifices some accuracy, and the divide-and-conquer strategy is not ideal for quantities that depend delicately on long-range effects, such as precise binding energies in tight protein–ligand complexes. The authors show that for such high-precision tasks the cutoffs must be relaxed or removed, and the method behaves more like a conventional, slower quantum code. Yet for very large systems where no fully accurate quantum treatment is feasible at all, their approach offers a powerful middle ground: it delivers qualitatively and often quantitatively reliable insights with modest computing resources, even down to single compute nodes for systems up to about a million atoms.
What This Means for the Future of Molecular Science
By re-engineering a standard quantum method to favor speed while staying in good agreement with experiments, this framework opens the door to routine quantum-level studies of entire viruses, protein machines and large DNA–drug complexes. It enables detailed electron-density maps that can be compared directly with cutting-edge crystallography, realistic optical spectra for biomolecules of unprecedented size and independent checks on AI-generated protein structures. Looking ahead, the same ideas could support fast quantum-based molecular dynamics, studies of how electric or magnetic fields influence biomolecules, and the generation of large, quantum-accurate datasets to train next-generation machine-learning models. In practical terms, it brings the once far-off dream of "quantum biology"—treating life’s machinery with full electronic detail—closer to everyday scientific practice.
Citation: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Keywords: quantum biomolecular simulation, Hartree–Fock, protein structure prediction, UV-Vis spectra, divide-and-conquer algorithms