Clear Sky Science · pl
Mechanika kwantowa dla biologicznych układów na poziomie milionów atomów
Zajrzeć w życie, elektron po elektronie
Białka, DNA i wirusy zawdzięczają swoje zachowanie nieustannemu ruchowi elektronów, jednak symulacje na prawdziwie kwantowym poziomie takich dużych cząsteczek długo były zbyt wolne, by stosować je poza najmniejszymi układami. Niniejsza praca przedstawia sposób na wprowadzenie pełnej mechaniki kwantowej do ogromnych struktur biologicznych — sięgających dziesiątek milionów atomów — przez ostrożne obniżenie dokładności tylko na tyle, by obliczenia stały się szybkie, przystępne i zaskakująco użyteczne dla pytań biologicznych, medycznych i związanych z projektowaniem leków.
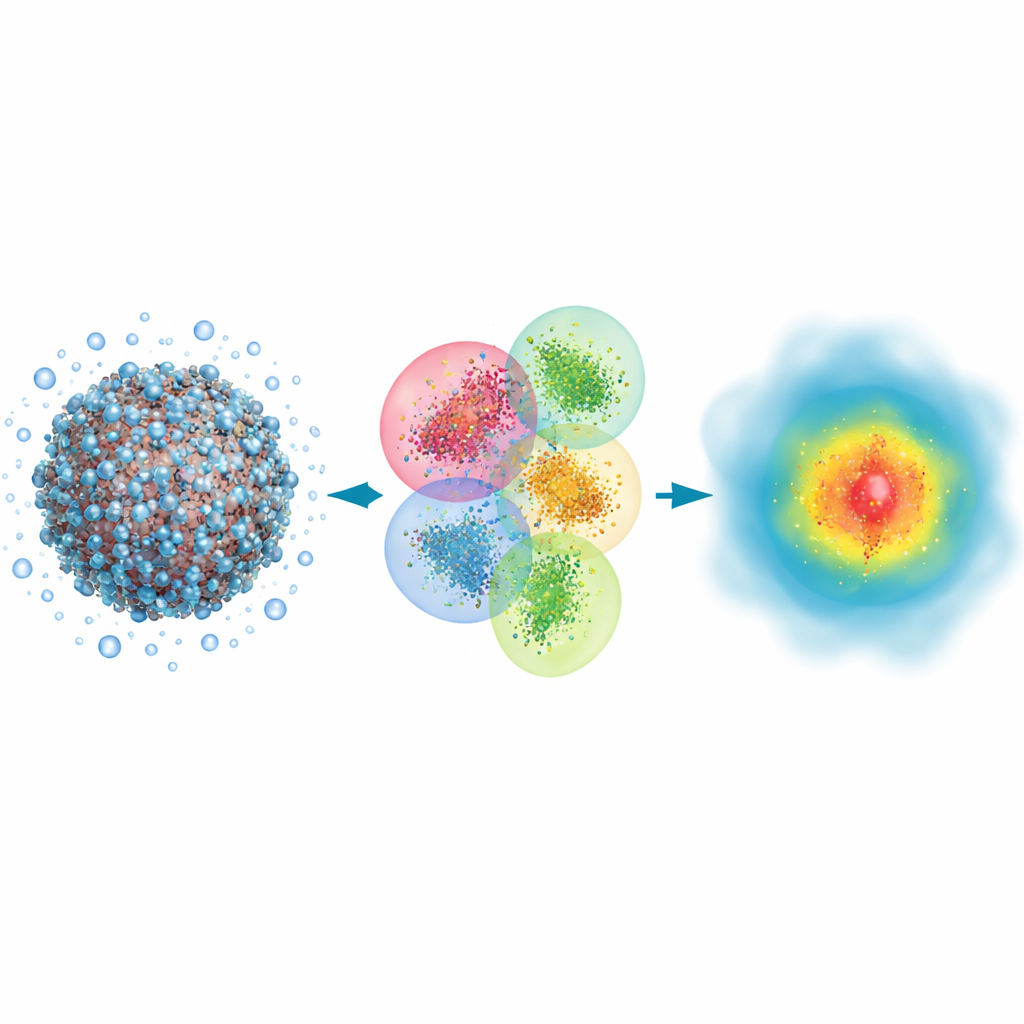
Dlaczego duże cząsteczki potrzebują kwantowego spojrzenia
Wiele procesów biologicznych, od wiązania leku po działanie światła w widzeniu, wiąże się z łamaniem i tworzeniem wiązań chemicznych oraz przemieszczaniem chmur elektronowych. Klasyczne metody symulacji potrafią śledzić ogólny ruch atomów w dużych białkach czy DNA, ale traktują elektrony jedynie pośrednio i pomijają subtelniejsze efekty, które decydują o tym, jak absorbuje się światło lub jak silnie lek przyczepia się do celu. Z kolei podejścia kwantowo-mechaniczne śledzą elektrony jawnie i potrafią przewidywać właściwości, takie jak widma optyczne, z wysoką wiernością. Problem w tym, że tradycyjne metody kwantowe zwalniają gwałtownie wraz ze wzrostem rozmiaru układu, więc zwykle stosowano je tylko do małych fragmentów, pozostawiając całe wirusy czy olbrzymie zespoły białkowe poza zasięgiem.
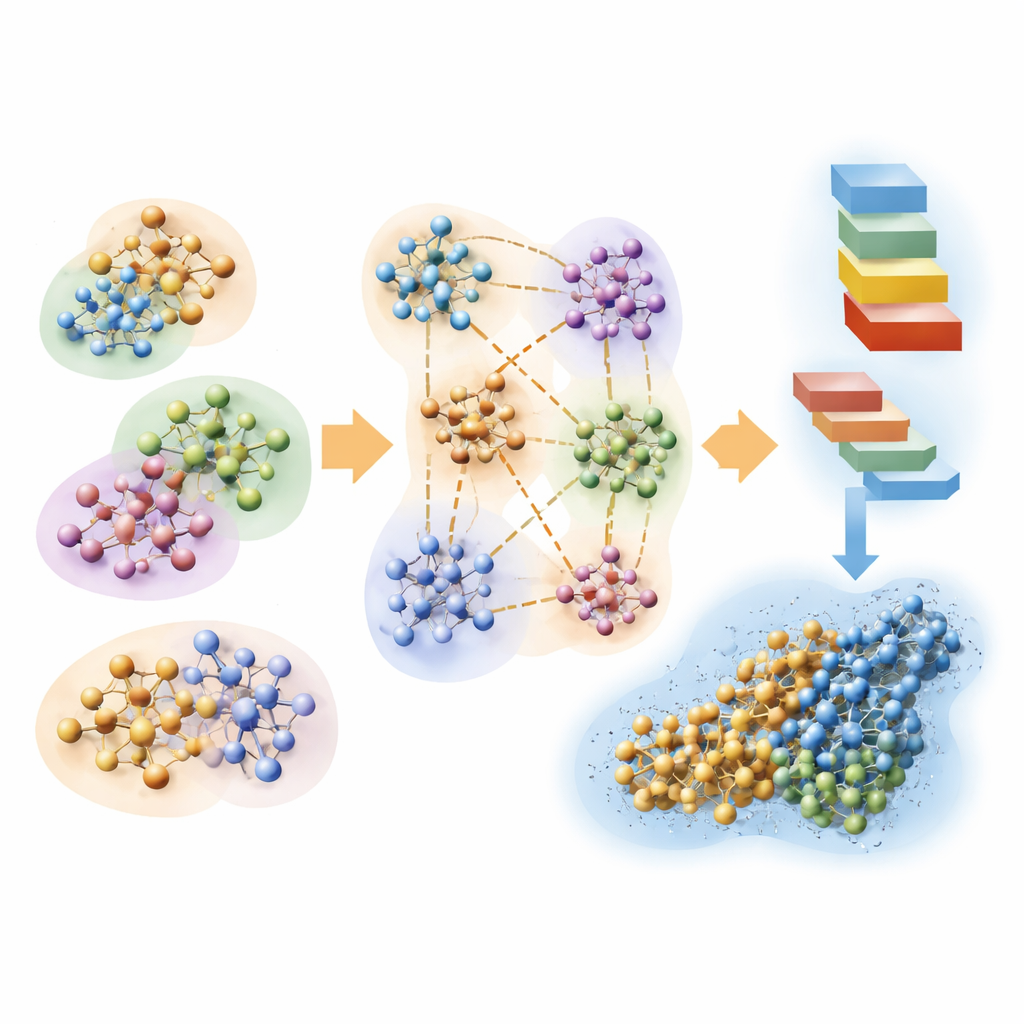
Podział problemu na gryzione kawałki
Autorzy bazują na klasycznym kwantowym podejściu zwanym metodą Hartree–Focka, które opisuje elektrony za pomocą orbitali matematycznych, i przebudowują je pod kątem szybkości zamiast maksymalnej precyzji. Ich kluczowy pomysł to podział ogromnej biomolekuły wraz z otaczającą ją wodą na wiele nakładających się klastrów. Każdy klaster ma centralny region rdzeniowy, w którym zachowywane są wyniki kwantowe, oraz region buforowy naśladujący wpływ sąsiednich atomów. Obliczenia kwantowe przeprowadza się osobno dla każdego klastra, a fragmenty składa razem, aby odtworzyć pełne zagęszczenie elektronowe układu. Dzięki zastosowaniu bardzo zwartej reprezentacji orbitali oraz odrzuceniu słabych, dalekosiężnych oddziaływań, które w niewielkim stopniu wpływają na wynik, metoda skaluje się w przybliżeniu proporcjonalnie do liczby atomów zamiast eksplodować kosztami.
Od wirusów po DNA i leki przeciwnowotworowe
Aby pokazać możliwości uproszczonego frameworku, badacze zastosowali go do kilku pokazowych układów. Obliczyli pełną strukturę elektronową trzech dużych zespołów w wodzie, w tym bakteriofaga — wirusa atakującego bakterie — wraz z otaczającą go skrzynką rozpuszczalnika obejmującą ponad 45 milionów atomów i ponad 150 milionów elektronów. Na nowoczesnym klastrze obliczenie to kończy się w około pół dnia, co w ich opinii stanowi największą symulację Hartree–Focka wykonaną do tej pory. Następnie przeszli do absorpcji światła: używając rozszerzenia czasowo-zależnego swojej metody, symulowali widma ultrafioletowo–widzialne dla fragmentów DNA do 21 par zasad oraz dla leku przeciwnowotworowego aktynomycyny D, zarówno wolnych, jak i związanych z DNA. Przewidywane widma dobrze pokrywają się z pomiarami eksperymentalnymi, pokazując, że schemat z obniżoną dokładnością nadal uchwycił istotną fizykę interakcji tych biomolekuł ze światłem.
Weryfikacja kształtów białek przewidzianych przez AI za pomocą fizyki
Zespół bada również, jak szybkie obliczenia kwantowe mogą pomóc ocenić struktury białek przewidziane przez narzędzia sztucznej inteligencji, takie jak AlphaFold. Białka naturalnie składają się w kształty minimalizujące ich energię. Korzystając z liniowo skalującej metody Hartree–Focka, autorzy oszacowali lokalne energie atomowe wzdłuż długości kilku białek przewidzianych przez AlphaFold, w realistycznym otoczeniu wodnym. Gdy wzorce energii pochodzące z obliczeń kwantowych porównano z ocenami pewności AlphaFold, okazały się one uderzająco zgodne: niskie energie pokrywają się z regionami uznanymi przez AlphaFold za wiarygodne, podczas gdy wysokie energie wskazują niepewne lub niestabilne fragmenty. Sugeruje to, że nowoczesne sieci predykcyjne struktury pośrednio uczą się aspektów leżącego u podstaw kwantowego krajobrazu energetycznego, a obliczenia z pierwszych zasad mogą dostarczyć niezależnej, opartej na fizyce kontroli jakości.
Kompro-misy, ograniczenia i nowe możliwości
Zyski prędkości wynikają ze świadomych kompromisów. Użycie minimalnej bazy orbitali i odcinanie słabych, odległych oddziaływań koniecznie kosztem pewnej dokładności, a strategia dziel i zwyciężaj nie jest optymalna dla wielkości zależnych delikatnie od efektów dalekiego zasięgu, takich jak precyzyjne energie wiązania w ciasnych kompleksach białko–ligand. Autorzy pokazują, że dla takich zadań wymagających wysokiej precyzji trzeba poluzować lub usunąć progi odcięcia, a metoda zaczyna zachowywać się bardziej jak konwencjonalny, wolniejszy kod kwantowy. Jednak dla bardzo dużych układów, gdzie żadna w pełni dokładna kwantowa analiza w ogóle nie jest wykonalna, ich podejście stanowi potężny kompromis: dostarcza jakościowo, a często ilościowo wiarygodnych wglądów przy umiarkowanych zasobach obliczeniowych, nawet na pojedynczych węzłach obliczeniowych dla układów do około miliona atomów.
Co to oznacza dla przyszłości nauki o molekułach
Przeprojektowując standardową metodę kwantową, by faworyzowała szybkość przy jednoczesnym zachowaniu dobrej zgodności z eksperymentem, ten framework otwiera drzwi do rutynowych badań na poziomie kwantowym całych wirusów, maszyn białkowych i dużych kompleksów DNA–lek. Umożliwia szczegółowe mapy gęstości elektronowej, które można bezpośrednio porównać z najnowocześniejszą krystalografią, realistyczne widma optyczne dla biomolekuł o bezprecedensowych rozmiarach oraz niezależne weryfikacje struktur generowanych przez AI. Patrząc w przyszłość, te same pomysły mogłyby wspierać szybkie kwantowe dynamiki molekularne, badania wpływu pól elektrycznych lub magnetycznych na biomolekuły oraz tworzenie dużych, kwantowo dokładnych zestawów danych do trenowania kolejnej generacji modeli uczenia maszynowego. W praktyce przybliża to dawny odległy sen „kwantowej biologii” — traktowania maszynerii życia z pełnymi szczegółami elektronowymi — do codziennej praktyki naukowej.
Cytowanie: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Słowa kluczowe: kwantowa symulacja biomolekularna, Hartree–Fock, predykcja struktury białek, widma UV-Vis, algorytmy dziel i zwyciężaj