Clear Sky Science · es
Un marco cuántico para sistemas biológicos a escala de millones de átomos
Mirando dentro de la vida, un electrón a la vez
Las proteínas, el ADN y los virus deben su comportamiento al movimiento constante de los electrones, pero las simulaciones verdaderamente a nivel cuántico de moléculas tan grandes han sido durante mucho tiempo demasiado lentas para aplicarse salvo en los sistemas más pequeños. Este trabajo presenta una forma de llevar la mecánica cuántica completa a estructuras biológicas enormes —hasta decenas de millones de átomos— relajando cuidadosamente la precisión lo justo para que los cálculos sean rápidos, asequibles y sorprendentemente útiles para preguntas en biología, medicina y diseño de fármacos.
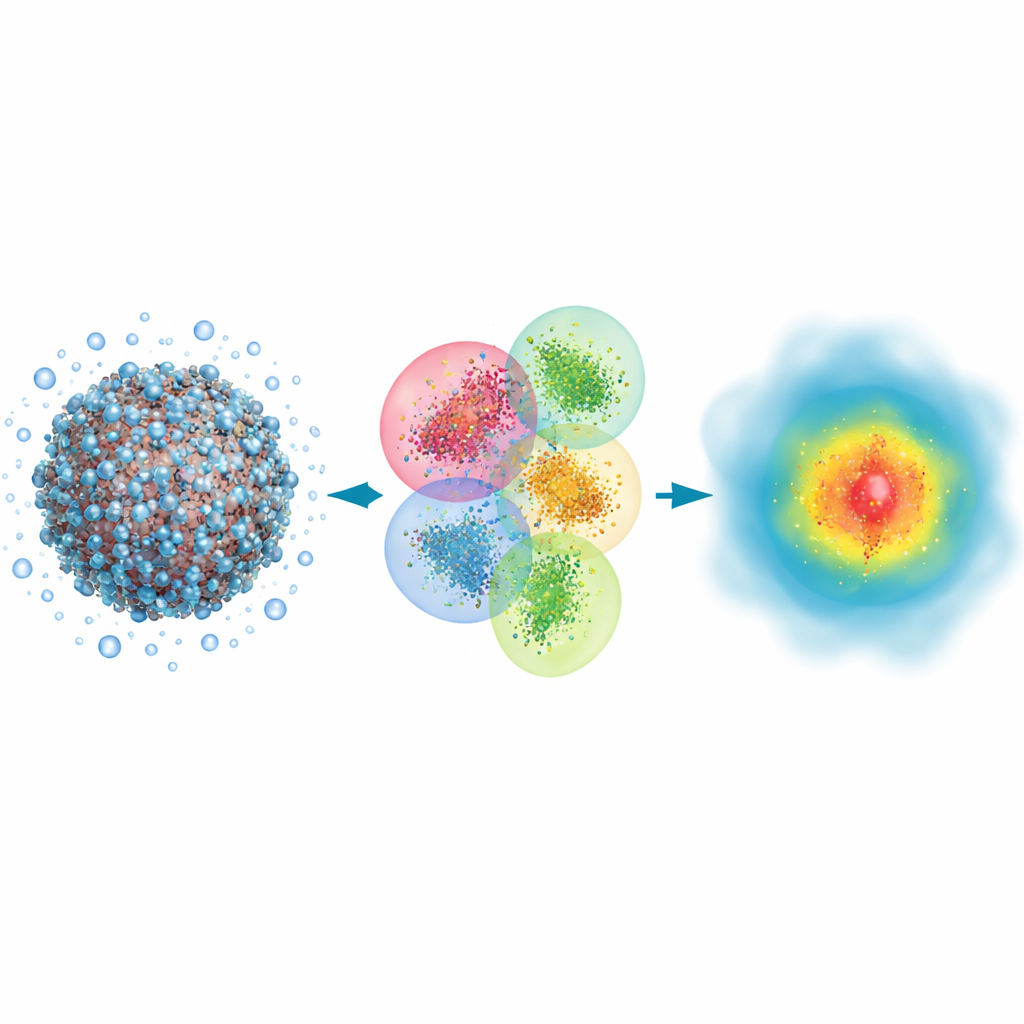
Por qué las moléculas grandes necesitan ojos cuánticos
Muchos eventos biológicos, desde la unión de un fármaco hasta la acción de la luz en la visión, implican romper y formar enlaces químicos y redistribuir las nubes electrónicas. Los métodos clásicos de simulación pueden seguir el movimiento general de los átomos en grandes proteínas o en el ADN, pero tratan a los electrones solo de forma indirecta y se pierden algunos de los efectos más sutiles que determinan cómo se absorbe la luz o con qué fuerza se adhiere un fármaco a su diana. En contraste, los enfoques cuántico-mecánicos siguen explícitamente a los electrones y pueden predecir propiedades como espectros ópticos con gran fidelidad. El inconveniente es que los métodos cuánticos tradicionales se vuelven muy lentos a medida que el sistema crece, por lo que normalmente se han usado solo para fragmentos pequeños, dejando enteros virus o enormes ensamblajes proteicos fuera de alcance.
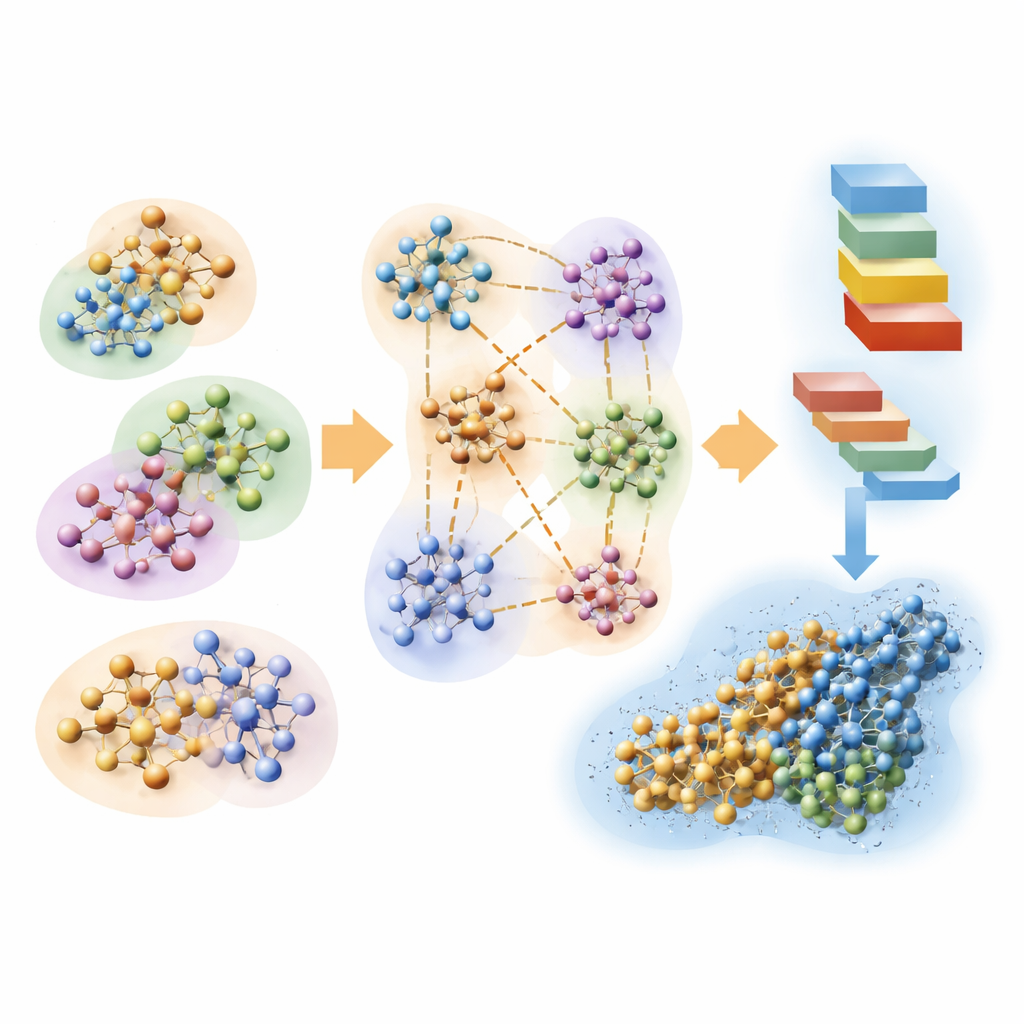
Dividir el problema en porciones manejables
Los autores se basan en un enfoque cuántico clásico llamado Hartree–Fock, que describe a los electrones mediante orbitales matemáticos, y lo reconfiguran para priorizar la velocidad sobre la máxima precisión. Su idea clave es dividir una biomolécula enorme más su agua circundante en muchos clústeres superpuestos. Cada clúster tiene una región central donde se conservan los resultados cuánticos y una región de contorno que imita la influencia de los átomos vecinos. A continuación se realizan cálculos cuánticos por separado en cada clúster y las piezas se ensamblan para reconstruir la densidad electrónica completa del sistema. Al usar además una representación muy compacta de los orbitales y descartar interacciones débiles y de largo alcance que contribuyen poco al resultado final, el método escala aproximadamente en proporción al número de átomos en lugar de explotar en coste.
De virus a ADN y fármacos contra el cáncer
Para demostrar lo que este marco optimizado puede hacer, los investigadores abordan varios sistemas emblemáticos. Calculan la estructura electrónica completa de tres grandes ensamblajes en agua, incluido un bacteriófago —un virus que infecta bacterias— junto con una caja de disolvente circundante que suma más de 45 millones de átomos y más de 150 millones de electrones. En un clúster moderno este cálculo termina en aproximadamente medio día, lo que representa, según su conocimiento, la mayor simulación Hartree–Fock jamás realizada. Luego se centran en la absorción de luz: usando una extensión dependiente del tiempo de su método, simulan espectros ultravioleta–visible para fragmentos de ADN de hasta 21 pares de bases y para el fármaco anticancerígeno Actinomicina D, tanto libre como unido al ADN. Los espectros predichos concuerdan estrechamente con las mediciones experimentales, mostrando que el esquema de precisión reducida aún captura la física esencial de cómo estas biomoléculas interactúan con la luz.
Comprobar las formas de proteínas predichas por IA con física
El equipo también explora cómo los cálculos cuánticos rápidos pueden ayudar a evaluar las estructuras de proteínas predichas por herramientas de inteligencia artificial como AlphaFold. Las proteínas se pliegan naturalmente en formas que minimizan su energía. Usando su enfoque Hartree–Fock de escalado lineal, los autores estiman energías atómicas locales a lo largo de varias proteínas predichas por AlphaFold, en entornos realistas de agua. Cuando estos patrones de energía derivados de la física cuántica se comparan con las propias puntuaciones de confianza de AlphaFold, coinciden de forma notable: las bajas energías se corresponden con regiones que AlphaFold considera fiables, mientras que las energías altas marcan segmentos inciertos o inestables. Esto sugiere que las modernas redes de predicción de estructuras están aprendiendo implícitamente aspectos del paisaje energético cuántico subyacente, y que los cálculos desde primeros principios pueden proporcionar una comprobación independiente basada en la física.
Compromisos, límites y nuevas posibilidades
Las ganancias de velocidad provienen de compromisos conscientes. Usar una base orbital mínima y cortar interacciones débiles y distantes sacrifica necesariamente algo de precisión, y la estrategia de dividir y conquistar no es ideal para cantidades que dependen delicadamente de efectos de largo alcance, como energías de unión precisas en complejos proteína–ligando muy ajustados. Los autores muestran que para tareas de alta precisión dichos recortes deben relajarse o eliminarse, y el método se comporta más como un código cuántico convencional y más lento. Sin embargo, para sistemas muy grandes en los que no es factible ningún tratamiento cuántico totalmente exacto, su enfoque ofrece un punto intermedio potente: aporta ideas cualitativas y, a menudo, cuantitativamente fiables con recursos informáticos modestos, incluso en nodos de cómputo individuales para sistemas de hasta aproximadamente un millón de átomos.
Lo que esto significa para el futuro de la ciencia molecular
Al reingeniar un método cuántico estándar para favorecer la velocidad sin perder un buen acuerdo con los experimentos, este marco abre la puerta a estudios de rutina a nivel cuántico de virus enteros, máquinas proteicas y grandes complejos ADN–fármaco. Permite mapas detallados de densidad electrónica que pueden compararse directamente con la cristalografía de vanguardia, espectros ópticos realistas para biomoléculas de tamaño sin precedentes y verificaciones independientes de estructuras proteicas generadas por IA. Mirando hacia el futuro, las mismas ideas podrían apoyar dinámica molecular rápida basada en la mecánica cuántica, estudios de cómo los campos eléctricos o magnéticos afectan a las biomoléculas y la generación de grandes conjuntos de datos con precisión cuántica para entrenar modelos de aprendizaje automático de próxima generación. En términos prácticos, acerca el sueño antaño lejano de la "biología cuántica" —tratar la maquinaria de la vida con detalle electrónico completo— más cerca de la práctica científica cotidiana.
Cita: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Palabras clave: simulación cuántica biomolecular, Hartree–Fock, predicción de estructuras de proteínas, espectros UV-Vis, algoritmos divide y vencerás