Clear Sky Science · it
Un quadro quantistico per sistemi biologici su scala di milioni di atomi
Scrutare la vita, un elettrone alla volta
Proteine, DNA e virus devono il loro comportamento al continuo movimento degli elettroni, ma simulazioni veramente quantistiche di molecole così grandi sono state a lungo troppo lente per essere eseguite se non su sistemi molto piccoli. Questo lavoro presenta un modo per portare la meccanica quantistica completa su strutture biologiche enormi — fino a decine di milioni di atomi — rilassando con attenzione la precisione il minimo necessario per rendere i calcoli veloci, economici e sorprendentemente utili per questioni in biologia, medicina e progettazione di farmaci.
Perché le grandi molecole hanno bisogno di occhi quantistici
Molti eventi biologici, dall’associazione di un farmaco all’azione della luce nella visione, implicano la rottura e la formazione di legami chimici e lo spostamento delle nuvole elettroniche. I metodi classici di simulazione possono seguire il moto complessivo degli atomi in grandi proteine o nel DNA, ma trattano gli elettroni in modo solo indiretto e perdono alcuni degli effetti più sottili che determinano come la luce viene assorbita o quanto saldamente un farmaco si lega al suo bersaglio. Al contrario, gli approcci quantistici trattano gli elettroni esplicitamente e possono prevedere proprietà come gli spettri ottici con alta fedeltà. Il problema è che i metodi quantistici tradizionali rallentano drasticamente all’aumentare della dimensione del sistema, quindi sono stati tipicamente usati solo per piccoli frammenti, lasciando interi virus o grandi assemblaggi proteici fuori portata.
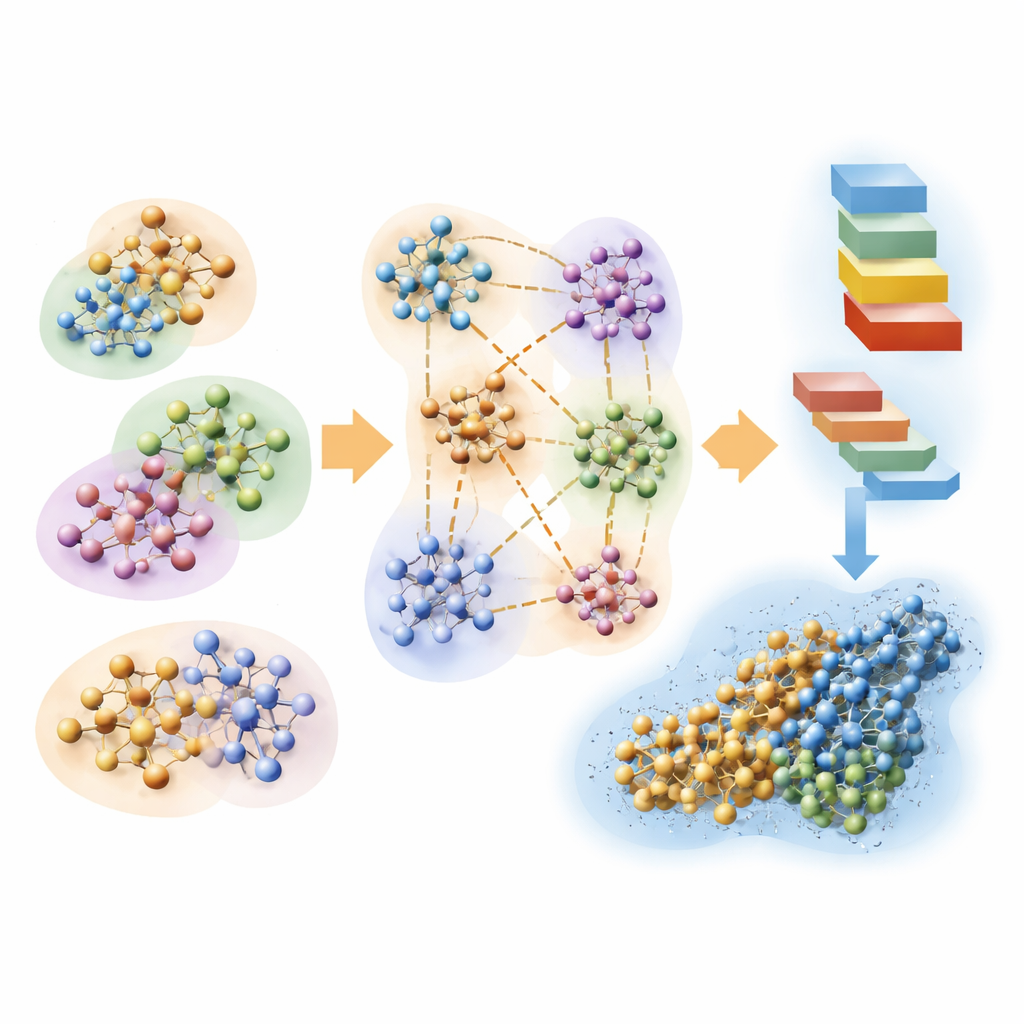
Spezzare il problema in pezzi gestibili
Gli autori partono da un approccio quantistico classico chiamato Hartree–Fock, che descrive gli elettroni con orbitali matematici, e lo rimodellano per la velocità più che per la precisione estrema. L’idea chiave è suddividere una grande biomolecola e la sua acqua circostante in molti cluster sovrapposti. Ogni cluster ha una regione centrale dove i risultati quantistici vengono mantenuti e una regione di buffer che imita l’influenza degli atomi vicini. I calcoli quantistici vengono quindi eseguiti separatamente su ciascun cluster e i pezzi vengono cuciti insieme per ricostruire la densità elettronica completa del sistema. Utilizzando inoltre una rappresentazione molto compatta degli orbitali e scartando interazioni deboli e a lunga distanza che contribuiscono poco al risultato finale, il metodo scala approssimativamente in proporzione al numero di atomi anziché esplodere in costo computazionale.
Da virus a DNA e farmaci antitumorali
Per dimostrare cosa può fare questo quadro semplificato, i ricercatori affrontano diversi sistemi di riferimento. Calcolano la struttura elettronica completa di tre grandi assemblaggi in acqua, incluso un batteriofago — un virus che infetta i batteri — insieme a una scatola di solvente che supera i 45 milioni di atomi e oltre 150 milioni di elettroni. Su un cluster moderno questo calcolo termina in circa mezza giornata, rappresentando, per quanto ne sappiano, la più grande simulazione Hartree–Fock mai eseguita. Passano poi all’assorbimento della luce: usando un’estensione tempo-dipendente del loro metodo, simulano spettri ultravioletto-visibili per frammenti di DNA fino a 21 coppie di basi e per il farmaco antitumorale Actinomicina D, sia libero sia legato al DNA. Gli spettri predetti corrispondono strettamente alle misure sperimentali, mostrando che lo schema a precisione ridotta cattura comunque la fisica essenziale di come queste biomolecole interagiscono con la luce.
Verificare le forme proteiche predette dall’IA con la fisica
Il gruppo esplora anche come i calcoli quantistici rapidi possano aiutare a valutare le strutture proteiche predette da strumenti di intelligenza artificiale come AlphaFold. Le proteine si ripiegano naturalmente in forme che minimizzano la loro energia. Usando il loro approccio Hartree–Fock a scala lineare, gli autori stimano le energie atomiche locali lungo diverse proteine predette da AlphaFold, in ambienti acquosi realistici. Quando questi pattern energetici di derivazione quantistica vengono confrontati con i punteggi di confidenza di AlphaFold, si allineano in modo sorprendentemente consistente: energie basse coincidono con regioni che AlphaFold giudica affidabili, mentre energie alte segnalano segmenti incerti o instabili. Questo suggerisce che le moderne reti di predizione strutturale stanno implicitamente apprendendo aspetti del paesaggio energetico quantistico sottostante, e che i calcoli di prima principio possono fornire un controllo di qualità indipendente e basato sulla fisica.
Compromessi, limiti e nuove possibilità
I guadagni di velocità derivano da compromessi consapevoli. Usare una base di orbitali minima e tagliare interazioni deboli e distanti sacrifica necessariamente parte della precisione, e la strategia divide-and-conquer non è ideale per quantità che dipendono delicatamente da effetti di lunga portata, come energie di legame precise in complessi proteina–ligando molto stretti. Gli autori mostrano che per compiti ad alta precisione i tagli devono essere rilassati o rimossi, e il metodo si comporta più come un codice quantistico convenzionale e più lento. Tuttavia, per sistemi molto grandi dove nessun trattamento quantistico pienamente accurato è fattibile, il loro approccio offre un potente compromesso: fornisce intuizioni qualitative e spesso quantitative affidabili con risorse computazionali modeste, anche su singoli nodi di calcolo per sistemi fino a circa un milione di atomi.
Cosa significa per il futuro della scienza molecolare
Riprogettando un metodo quantistico standard per privilegiare la velocità mantenendo un buon accordo con gli esperimenti, questo quadro apre la porta a studi di routine a livello quantistico su interi virus, macchine proteiche e grandi complessi DNA–farmaco. Permette mappe dettagliate della densità elettronica confrontabili direttamente con la cristallografia d’avanguardia, spettri ottici realistici per biomolecole di dimensioni senza precedenti e controlli indipendenti sulle strutture proteiche generate dall’IA. Guardando avanti, le stesse idee potrebbero sostenere dinamiche molecolari quantistiche rapide, studi su come campi elettrici o magnetici influenzano le biomolecole e la generazione di grandi dataset quantisticamente accurati per addestrare modelli di apprendimento automatico di nuova generazione. In termini pratici, avvicina il sogno una volta lontano della “biologia quantistica” — trattare i meccanismi della vita con il dettaglio elettronico completo — alla pratica scientifica quotidiana.
Citazione: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Parole chiave: simulazione quantistica biomolecolare, Hartree–Fock, predizione della struttura proteica, spettri UV-Vis, algoritmi divide-and-conquer