Clear Sky Science · de
Ein quantenmechanischer Rahmenwerk für biologische Systeme im Million-Atom-Maßstab
Ins Leben schauen, ein Elektron nach dem anderen
Proteine, DNA und Viren verdanken ihr Verhalten der unruhigen Bewegung von Elektronen, doch wirklich quantenmechanische Simulationen so großer Moleküle waren lange Zeit nur für sehr kleine Systeme praktikabel. Diese Arbeit stellt einen Weg vor, vollständige Quantenmechanik auf gewaltige biologische Strukturen anzuwenden — bis hin zu mehreren zehn Millionen Atomen — indem die Genauigkeit gezielt nur so weit reduziert wird, dass die Rechnungen schnell, erschwinglich und überraschend nützlich für Fragestellungen in Biologie, Medizin und Wirkstoffdesign bleiben.
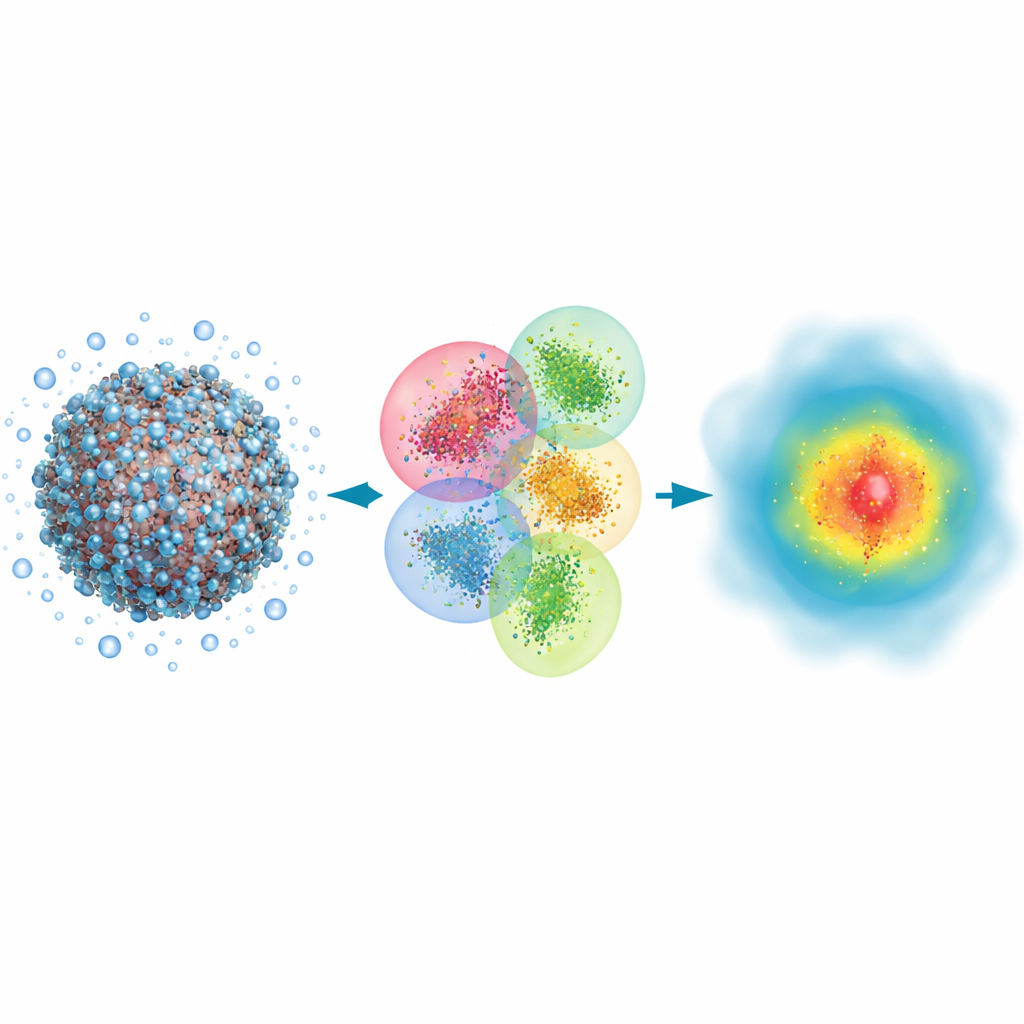
Warum große Moleküle Quantenblicke brauchen
Viele biologische Vorgänge, von der Wirkstoffbindung bis zur Wirkung des Sonnenlichts auf das Sehen, beinhalten das Brechen und Bilden chemischer Bindungen und das Verschieben von Elektronenwolken. Klassische Simulationsmethoden können die Gesamtbewegung von Atomen in großen Proteinen oder DNA nachverfolgen, behandeln Elektronen jedoch nur indirekt und übersehen einige feinere Effekte, die bestimmen, wie Licht absorbiert wird oder wie fest ein Wirkstoff an sein Ziel bindet. Im Gegensatz dazu verfolgen quantenmechanische Ansätze Elektronen explizit und können Eigenschaften wie optische Spektren mit hoher Treffsicherheit vorhersagen. Der Haken ist, dass traditionelle Quantenmethoden mit wachsender Systemgröße drastisch langsamer werden, weshalb sie typischerweise nur für kleine Fragmente eingesetzt wurden und ganze Viren oder riesige Proteinassemblies außerhalb ihrer Reichweite blieben.
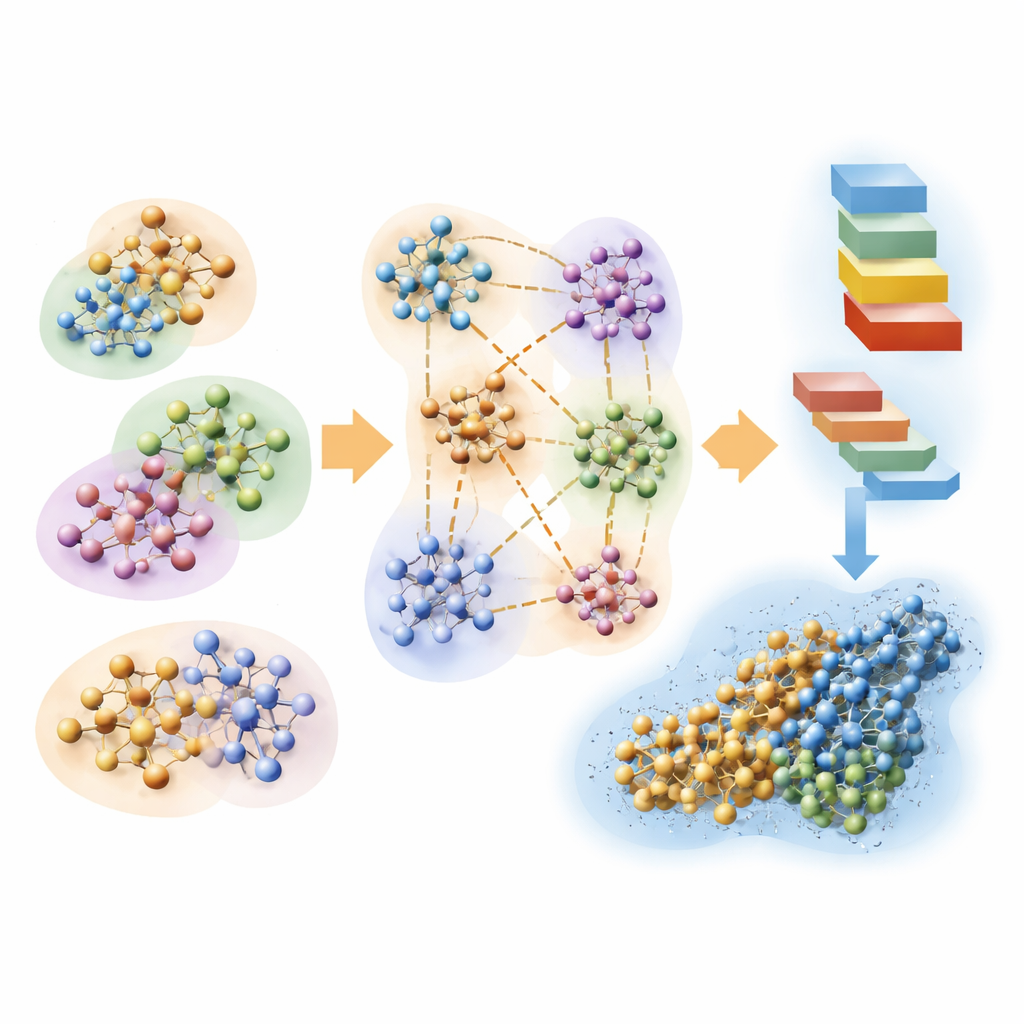
Das Problem in mundgerechte Stücke zerlegen
Die Autoren bauen auf einem klassischen Quantenansatz namens Hartree–Fock auf, der Elektronen durch mathematische Orbitale beschreibt, und formen ihn zugunsten von Geschwindigkeit statt ultimativer Präzision um. Ihre Schlüsselidee ist, ein riesiges Biomolekül plus seine umgebenden Wassermoleküle in viele überlappende Cluster zu teilen. Jeder Cluster hat einen zentralen Kernbereich, dessen quantenmechanische Ergebnisse beibehalten werden, und eine Pufferzone, die den Einfluss benachbarter Atome nachahmt. Auf jedem Cluster werden dann getrennte Quantenberechnungen durchgeführt, und die Teile werden wieder zusammengesetzt, um die vollständige Elektronendichte des Systems zu rekonstruieren. Durch die Verwendung einer sehr kompakten Darstellung der Orbitale und das Vernachlässigen schwacher, langreichweitiger Wechselwirkungen, die wenig zum Endergebnis beitragen, skaliert die Methode grob proportional zur Anzahl der Atome, anstatt in den Kosten exponentiell zu explodieren.
Von Viren zu DNA und Krebsmedikamenten
Um zu zeigen, was dieses gestraffte Framework leisten kann, bearbeiten die Forscher mehrere repräsentative Systeme. Sie berechnen die vollständige elektronische Struktur dreier großer Assemblies in Wasser, darunter ein Bakteriophagen — ein Virus, das Bakterien infiziert — zusammen mit einer umgebenden Lösungskiste mit mehr als 45 Millionen Atomen und über 150 Millionen Elektronen. Auf einem modernen Cluster ist diese Rechnung in etwa einem halben Tag abgeschlossen und stellt nach ihrem Wissen die größte jemals durchgeführte Hartree–Fock-Simulation dar. Anschließend wenden sie sich der Lichtabsorption zu: Mit einer zeitabhängigen Erweiterung ihrer Methode simulieren sie Ultraviolett–Sichtbar-Spektren für DNA-Fragmente bis zu 21 Basenpaaren und für das krebsbekämpfende Medikament Actinomycin D, sowohl frei als auch an DNA gebunden. Die vorhergesagten Spektren stimmen eng mit experimentellen Messungen überein und zeigen, dass das Genauigkeitsreduktionsschema dennoch die wesentliche Physik erfasst, wie diese Biomoleküle mit Licht interagieren.
KI-vorhergesagte Proteinfalten physikalisch prüfen
Das Team untersucht außerdem, wie schnelle Quantenberechnungen helfen können, Proteinstrukturen zu bewerten, die von KI-Werkzeugen wie AlphaFold vorhergesagt wurden. Proteine falten sich natürlicherweise in Formen, die ihre Energie minimieren. Mithilfe ihres linear skalierenden Hartree–Fock-Ansatzes schätzen die Autoren lokale Atomenergien entlang der Länge mehrerer AlphaFold-vorhergesagter Proteine in realistischen Wasserumgebungen ab. Wenn diese quantenbasierten Energieprofile mit AlphaFolds eigenen Vertrauenswerten verglichen werden, stimmen sie auffallend gut überein: niedrige Energien fallen mit Bereichen zusammen, die AlphaFold als zuverlässig einstuft, während hohe Energien unsichere oder instabile Segmente markieren. Das deutet darauf hin, dass moderne Strukturvorhersagenetzwerke implizit Aspekte der zugrundeliegenden quantenmechanischen Energielandschaft erlernen und dass Erstprinzipien-Berechnungen eine unabhängige, physikbasierte Qualitätsprüfung liefern können.
Abwägungen, Grenzen und neue Möglichkeiten
Die Geschwindigkeitsgewinne beruhen auf bewussten Kompromissen. Die Verwendung einer minimalen Orbitalbasis und das Abschneiden schwacher, entfernter Wechselwirkungen opfern zwangsläufig etwas Genauigkeit, und die Teile-und-herrsche-Strategie ist nicht ideal für Größen, die empfindlich von langreichweitigen Effekten abhängen, wie etwa präzise Bindungsenergien in engen Protein–Ligand-Komplexen. Die Autoren zeigen, dass für solche hochpräzisen Aufgaben die Abschneidewerte gelockert oder aufgehoben werden müssen und die Methode sich dann eher wie ein konventioneller, langsamerer Quantencode verhält. Dennoch bietet ihr Ansatz für sehr große Systeme, für die keine vollständig genaue Quantenbehandlung überhaupt möglich ist, einen mächtigen Mittelweg: Er liefert qualitativ und oft auch quantitativ verlässliche Einsichten mit moderaten Rechenressourcen, bis hin zu einzelnen Rechenknoten für Systeme bis zu etwa einer Million Atome.
Was das für die Zukunft der Molekularwissenschaft bedeutet
Indem ein standardmäßiges Quantenverfahren neu konzipiert wird, um Geschwindigkeit zu favorisieren und gleichzeitig in guter Übereinstimmung mit Experimenten zu bleiben, eröffnet dieses Framework die Tür zu routinemäßigen quantenmechanischen Studien ganzer Viren, Proteinmaschinen und großer DNA–Wirkstoff-Komplexe. Es ermöglicht detaillierte Elektronendichtemaps, die direkt mit moderner Kristallographie verglichen werden können, realistische optische Spektren für Biomoleküle bislang unerreichter Größe und unabhängige Prüfungen von KI-generierten Proteinstrukturen. In die Zukunft gedacht könnten dieselben Ideen schnelle quantenbasierte molekulare Dynamik, Studien darüber, wie elektrische oder magnetische Felder Biomoleküle beeinflussen, und die Erzeugung großer, quantengenauer Datensätze zur Schulung der nächsten Generation von Machine-Learning-Modellen unterstützen. Praktisch bringt es den einst fern erscheinenden Traum der „Quantenbiologie“ — die Behandlung von Lebensmaschinen mit vollständiger elektronischer Detailtreue — näher an die alltägliche wissenschaftliche Praxis.
Zitation: Wieners, L., Garcia, M.E. A quantum-mechanical framework for million-atom scale biological systems. Commun Chem 9, 170 (2026). https://doi.org/10.1038/s42004-026-02038-y
Schlüsselwörter: quanten-biomolekulare Simulation, Hartree–Fock, Proteinstrukturvorhersage, UV-Vis-Spektren, Teile-und-herrsche-Algorithmen