Clear Sky Science · sv
TH5487 riktar sig specifikt mot NLRP3 hos FCAS‑patienter som är resistenta mot MCC950
Varför det är viktigt att dämpa okontrollerad inflammation
Många kroniska sjukdomar, från sällsynta febersyndrom till åldersrelaterade åkommor, drivs av ett immunsystemets larm som vägrar stänga av. Denna studie undersöker nya läkandekandidater som kan tysta en av kroppens kraftfullaste inflammatoriska strömbrytare, kallad NLRP3, utan att helt slå ut vårt försvar. Genom att ompröva befintliga molekyler som ursprungligen riktades mot DNA‑reparation hittar författarna ett nytt sätt att tygla skadlig inflammation och till och med omdirigera den mot ett mer skyddande antiviral‑liknande svar.
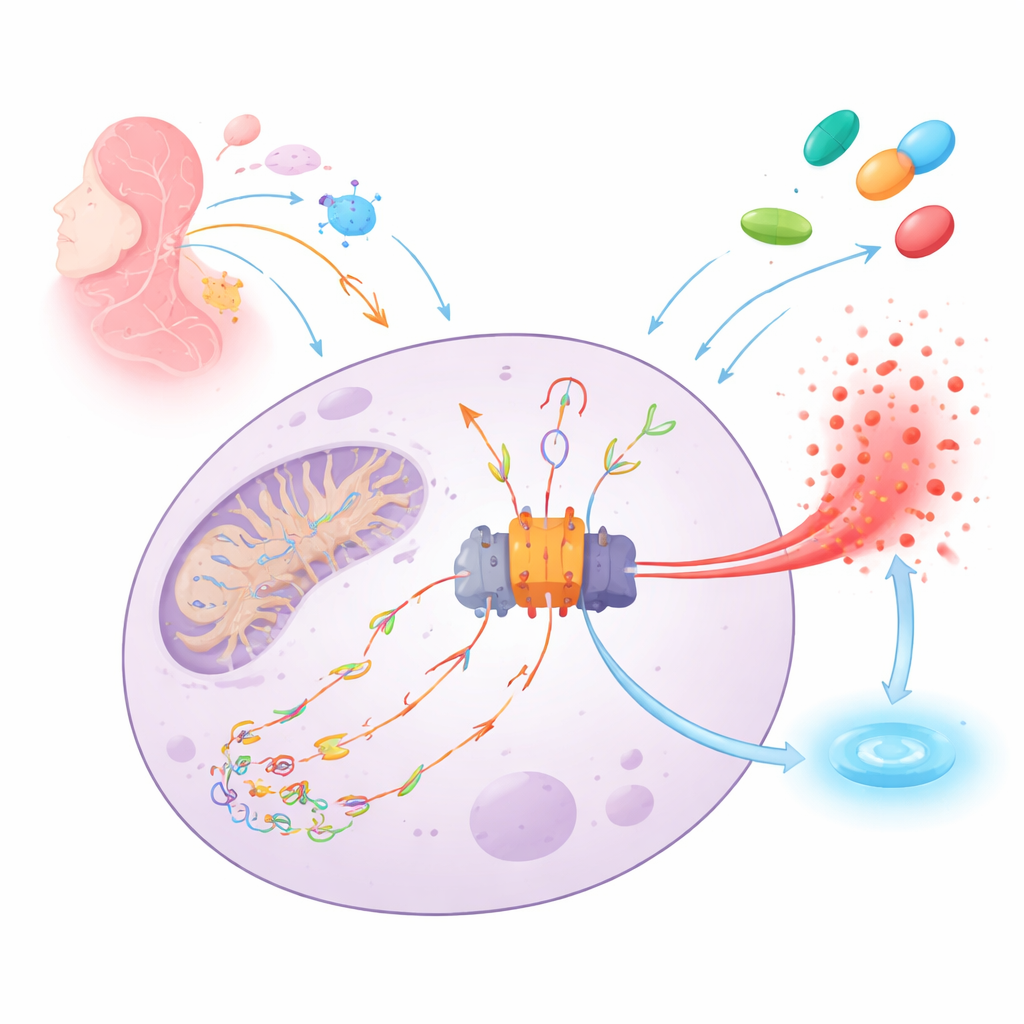
Kroppens brandlarm och dess gnista
NLRP3 fungerar som en molekylär rökdetektor inne i immunceller. När den känner av fara – allt från infektioner till vävnadsskada – monterar den ett stort proteinkomplex (inflammasomet) som utlöser frisättning av det potenta larmsignalen IL‑1β och kan få cellen att brista för att varna sina grannar. En viktig gnista för detta larm är skadat mitokondrie‑DNA som nickats och oxiderats av reaktiva syreföreningar. Dessa skadade DNA‑fragment kan läcka ut från mitokondrierna, vandra genom cellen, binda NLRP3 och driva montering av inflammasomet och inflammatorisk celldöd.
Gamla DNA‑reparationsläkemedel, nya antiinflammatoriska roller
Författarna undersökte små molekyler som först designades för att rikta in sig på ett DNA‑reparationsenzym kallat hOGG1, vilket avlägsnar oxiderade baser från DNA. Överraskande nog interagerar dessa läkemedel – TH5487, SU0268 och en aktivator benämnd TH10785 – också med NLRP3. I mänskliga blodimmunceller och modellcellinjer minskade låga mikromolära doser av TH5487 och SU0268 kraftigt frisättningen av IL‑1β och aktiveringen av caspas‑1, enzymet som mognar denna cytokin. Detaljerade protein pull‑down‑ och bildstudier visade att läkemedlen förhindrar NLRP3 från att montera sitt fullständiga inflammasomkomplex: de blockerar rekryteringen av viktiga partner (ASC, NEK7 och pro‑caspas‑1) och minskar bildandet av de ljusa ”prickarna” som kännetecknar ett aktivt inflammasom.
Klipper länken mellan skadat DNA och inflammasomet
Eftersom mitokondrieskada är en huvuddrivkraft för NLRP3‑aktivitet testade forskarna om läkemedlen kunde blockera inflammasomaktivering som utlöses av mitokondriell stress. En förening som stör cellens energifabriker och ökar reaktiva syreföreningar skickar normalt oxiderat mitokondrie‑DNA mot NLRP3 och höjer kraftigt IL‑1β‑nivåerna. TH5487 och SU0268 minskade inte bara IL‑1β‑frisättningen under dessa förhållanden, de reducerade också mängden NLRP3 som lokaliserades vid mitokondrier. Kryo‑elektronmikroskopi bekräftade att NLRP3 kan bilda ett stort ringformat komplex som fysiskt binder mitokondrie‑DNA, och datormodellering antydde att proteinets pyrindomän känner igen oxiderat enkelsträngat DNA via en positivt laddad yta. Läkemedlen stör detta DNA‑känslingssteg och bryter en avgörande koppling mellan skadat DNA och okontrollerad inflammation.
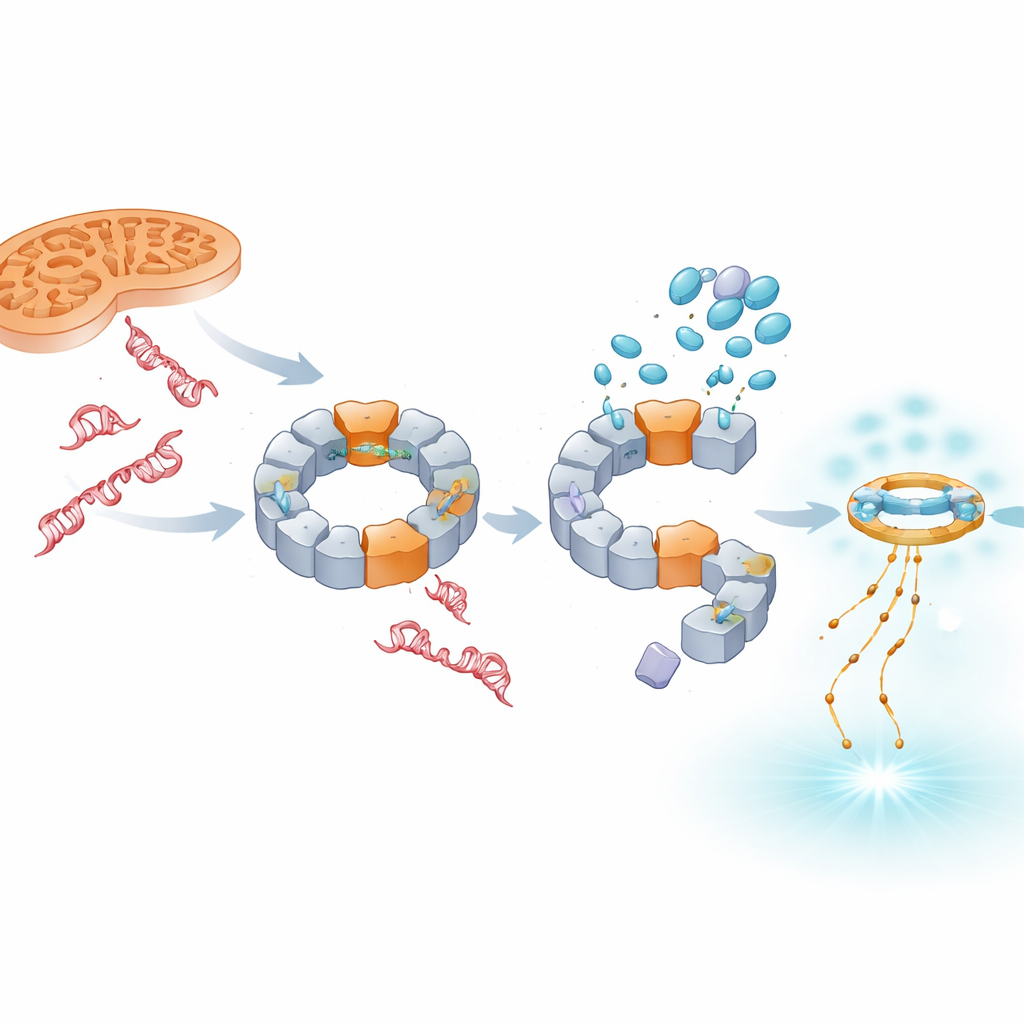
Förskjutning av signalen mot antiviral‑liknande försvar
Att blockera NLRP3 får inte det skadade DNA:t att försvinna; istället ackumuleras mer av det, särskilt från kärnan, i cellens cytosol. Studien visar att detta överskott av DNA plockas upp av ett annat sensorsystem, cGAS–STING, som typiskt reagerar på virusgenom. När TH5487, SU0268 eller TH10785 är närvarande ökar nivåerna av aktiverad STING och det antivirala budskapet IFN‑β i både modellceller och primära mänskliga blodceller. Viktigt är att denna förstärkning sker även när NLRP3 genetiskt tas bort, vilket indikerar att cGAS–STING fungerar som ett oberoende reservlarm. Således dämpar dessa föreningar IL‑1β‑driven inflammation samtidigt som de lutar immunsvaret mot interferonbaserad signalering som kan vara mer skyddande vid kronisk sjukdom.
Hopp för svårbehandlad ärftlig inflammation
Ett särskilt slående test rörde Familjär Kall Autoinflammatorisk Syndrom (FCAS), en sällsynt störning där mutationer i NLRP3 gör den hyperaktiv och resistent mot en ledande experimentell hämmare, MCC950. I immunceller från möss och mänskliga patienter med dessa mutationer lyckades inte MCC950 dämpa IL‑1β‑frisättningen. TH5487 och SU0268 däremot reducerade signifikant denna överproduktion vid doser som bevarade cellens livskraft. Detta tyder på att inriktning mot NLRP3:s oxiderade DNA‑kännande yta, snarare än dess mer traditionella enzymatiska kärna, kan kringgå resistens och erbjuda nya behandlingsalternativ för patienter vars sjukdom i dag har begränsade riktade terapier.
Vad dessa fynd betyder för framtida terapier
För en icke‑specialist är kärnbudskapet att forskarna funnit en ny ”avstängningsknapp” för ett kraftfullt inflammatoriskt maskineri inne i våra celler. Genom att återanvända DNA‑reparationsläkemedel för att blockera hur NLRP3 känner av skadat mitokondrie‑ och kärn‑DNA kan de dämpa en viktig källa till kronisk inflammation samtidigt som en annan väg får ta över och producera antivirala interferonsignaler. Om denna strategi visar sig vara säker i vidare studier kan den öppna för riktade behandlingar inte bara för sällsynta ärftliga febersyndrom som FCAS, utan även för bredare tillstånd av ”inflammaging” där persistenta, låggradiga inflammationsprocesser driver sjukdom.
Citering: Lackner, A., Picucci, S.I., Jiang, W. et al. TH5487 specifically targets NLRP3 in FCAS patients resistant to MCC950. Commun Biol 9, 528 (2026). https://doi.org/10.1038/s42003-026-10008-2
Nyckelord: NLRP3‑inflammasom, oxiderat mitokondrie‑DNA, TH5487, cGAS‑STING‑vägen, autoinflammatorisk sjukdom