Clear Sky Science · de
TH5487 zielt spezifisch auf NLRP3 bei FCAS-Patienten, die gegen MCC950 resistent sind
Warum es wichtig ist, außer Kontrolle geratene Entzündung zu beruhigen
Viele chronische Erkrankungen — von seltenen Fiebern bis zu altersbedingten Störungen — werden von einem Immunsystem‑Alarm getrieben, der sich nicht ausschaltet. Diese Studie untersucht neue Wirkstoffkandidaten, die einen der stärksten entzündlichen Schalter des Körpers, NLRP3, dämpfen können, ohne die Abwehr komplett auszuschalten. Indem die Autoren vorhandene Moleküle, die ursprünglich auf die DNA‑Reparatur abzielten, neu denken, zeigen sie einen frischen Weg auf, schädliche Entzündungen zu zügeln und sie sogar in eine schützende, antiviralartige Antwort umzulenken.
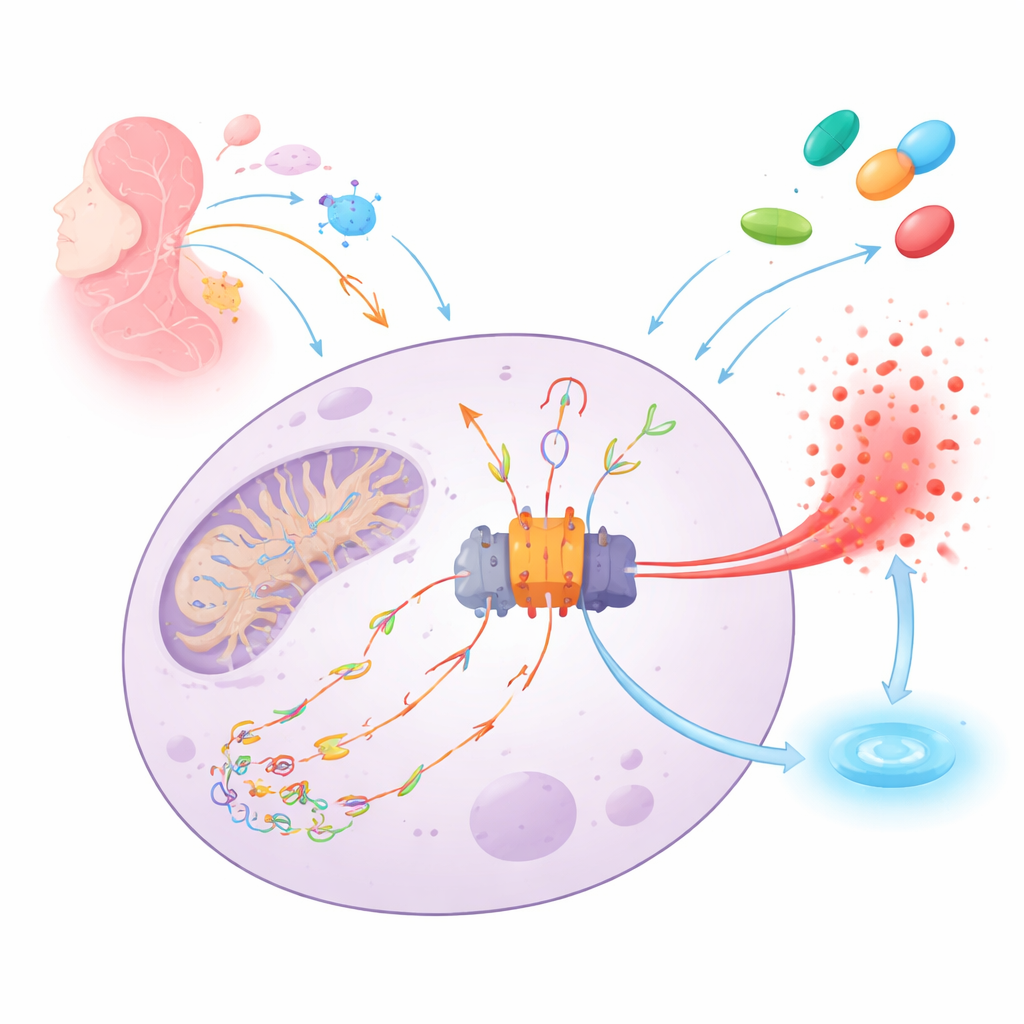
Der Feueralarm des Körpers und sein Funke
NLRP3 funktioniert wie ein molekularer Rauchmelder in Immunzellen. Wenn es Gefahr wahrnimmt — von Infektionen bis zu Gewebeschäden — setzt es ein großes Protein‑Gebilde (das Inflammasom) zusammen, das die Freisetzung des starken Alarmsignals IL‑1β auslöst und die Zelle zum Platzen bringen kann, um Nachbarzellen zu warnen. Ein zentraler Funke für diesen Alarm sind beschädigte mitochondriale DNA‑Fragmente, die durch reaktive Sauerstoffspezies aufgebrochen und oxidiert wurden. Diese beschädigten DNA‑Fragmente können aus den Mitochondrien entweichen, durch die Zelle wandern, an NLRP3 binden und die Assembly des Inflammasoms sowie den entzündlichen Zelltod antreiben.
Alte DNA‑Reparaturmedikamente, neue anti‑entzündliche Rollen
Die Autoren untersuchten kleine Moleküle, die ursprünglich entwickelt wurden, um ein DNA‑Reparaturenzym namens hOGG1 anzugreifen, das oxidierte Basen aus der DNA entfernt. Überraschenderweise interagieren diese Wirkstoffe — TH5487, SU0268 und ein Aktivator namens TH10785 — auch mit NLRP3. In humanen Blutzellen und Modellzelllinien reduzierten niedrige mikromolare Dosen von TH5487 und SU0268 deutlich die IL‑1β‑Freisetzung und die Aktivierung von Caspase‑1, dem Enzym, das dieses Zytokin reift. Detaillierte Protein‑Pull‑down‑ und Bildgebungs‑Experimente zeigten, dass die Wirkstoffe verhindern, dass NLRP3 sein vollständiges Inflammasom‑Komplex zusammenbaut: Sie blockieren die Rekrutierung zentraler Partner (ASC, NEK7 und Pro‑Caspase‑1) und verringern die Bildung der hellen „Specks“, die ein aktives Inflammasom kennzeichnen.
Die Verbindung zwischen beschädigter DNA und dem Inflammasom durchtrennen
Da mitochondriale Schäden ein wichtiger Treiber der NLRP3‑Aktivität sind, prüfte das Team, ob die Wirkstoffe die durch mitochondriale Belastung ausgelöste Inflammasom‑Aktivierung blockieren können. Eine Verbindung, die die Energieproduzenten der Zelle stört und reaktive Sauerstoffspezies erhöht, leitet normalerweise oxidierte mitochondriale DNA zu NLRP3 und steigert stark die IL‑1β‑Werte. TH5487 und SU0268 reduzierten unter diesen Bedingungen nicht nur die IL‑1β‑Freisetzung, sondern verringerten auch die Menge an NLRP3, die an Mitochondrien lokalisiert ist. Kryo‑Elektronenmikroskopie bestätigte, dass NLRP3 einen großen ringförmigen Komplex bilden kann, der physikalisch an mitochondriale DNA bindet, und Computermodelle deuteten darauf hin, dass die Pyrin‑Domäne des Proteins oxidierte einzelsträngige DNA über eine positiv geladene Oberflächenregion erkennt. Die Wirkstoffe stören diesen DNA‑Sensorschritt und durchtrennen damit eine entscheidende Verbindung zwischen beschädigter DNA und außer Kontrolle geratener Entzündung.
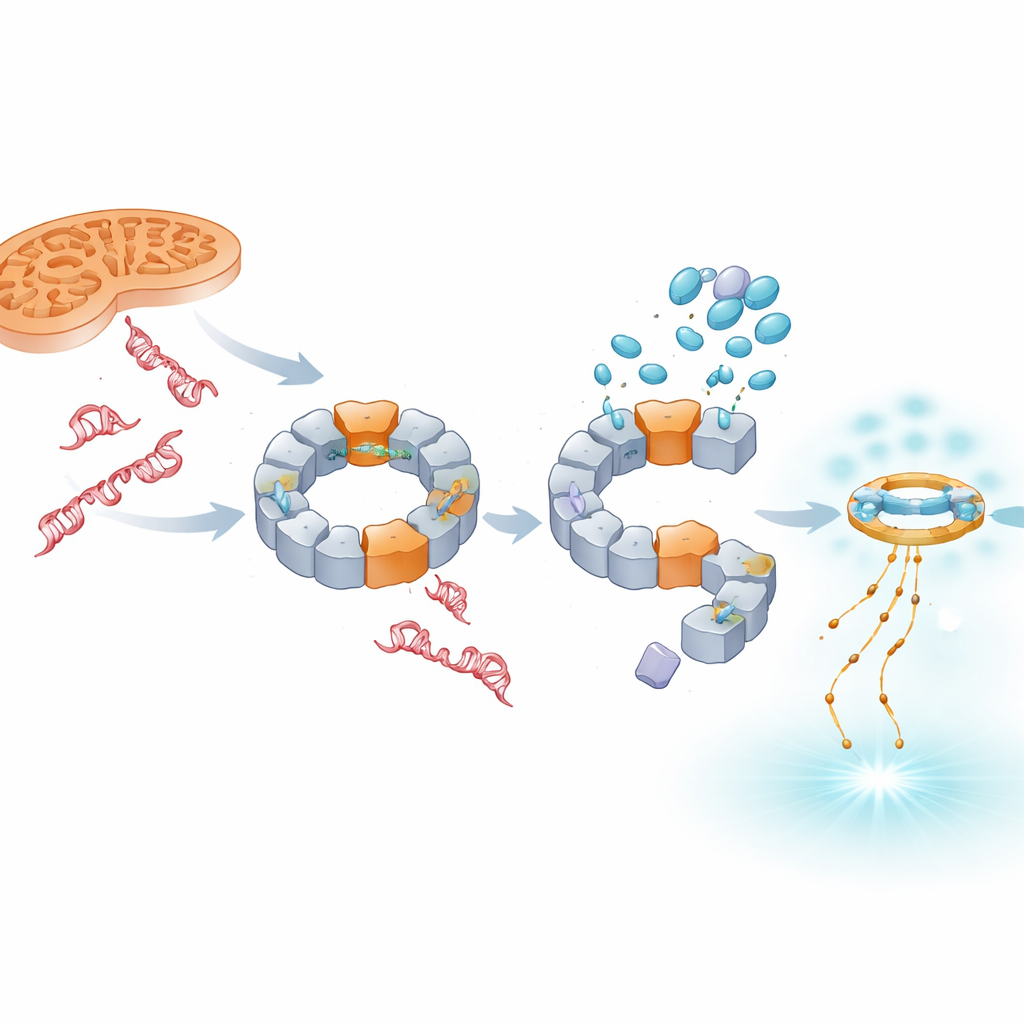
Das Signal in Richtung antiviral‑ähnlicher Abwehr verschieben
Das Blockieren von NLRP3 lässt die beschädigte DNA nicht verschwinden; stattdessen häuft sich mehr davon, insbesondere aus dem Zellkern, im Zellplasma an. Die Studie zeigt, dass diese überschüssige DNA von einem anderen Sensorsystem, cGAS–STING, aufgegriffen wird, das typischerweise auf virale Genome reagiert. Wenn TH5487, SU0268 oder TH10785 vorhanden sind, steigen die Aktivierung von STING und der Spiegel des antiviralen Boten IFN‑β sowohl in Modellzellen als auch in primären humanen Blutzellen. Wichtig ist, dass dieser Anstieg auch dann auftritt, wenn NLRP3 genetisch entfernt wurde, was darauf hinweist, dass cGAS–STING als unabhängiger Backup‑Alarm fungiert. Damit dämpfen diese Verbindungen IL‑1β‑getriebene Entzündung, während sie die Immunantwort in Richtung Interferon‑basierter Signalgebung neigen, die bei chronischen Erkrankungen möglicherweise schützender ist.
Versprechen für schwer behandelbare erbliche Entzündungen
Ein besonders eindrücklicher Test betraf das Familiäre Kälte‑Autoinflammatorische Syndrom (FCAS), eine seltene Erkrankung, bei der Mutationen in NLRP3 das Protein hyperaktiv machen und gegen einen führenden experimentellen Inhibitor, MCC950, resistent sind. In Immunzellen von Mäusen und menschlichen Patienten mit diesen Mutationen konnte MCC950 die IL‑1β‑Freisetzung nicht eindämmen. TH5487 und SU0268 hingegen reduzierten diese Überproduktion signifikant bei Dosen, die die Zellviabilität schonten. Das deutet darauf hin, dass die gezielte Ansprache der oxidierten DNA‑Erkennungsfläche von NLRP3 statt seines traditionelleren enzymatischen Kerns die Resistenz umgehen und neue Behandlungsoptionen für Patienten eröffnen könnte, deren Krankheit derzeit nur begrenzte zielgerichtete Therapien hat.
Was diese Ergebnisse für zukünftige Therapien bedeuten
Für Nicht‑Spezialisten lautet die Kernbotschaft, dass die Forscher einen neuen „Aus‑Schalter“ für eine potente entzündliche Maschine in unseren Zellen entdeckt haben. Indem sie DNA‑Reparaturmedikamente umnutzen, um zu blockieren, wie NLRP3 beschädigte mitochondriale und nukleäre DNA erkennt, können sie eine wichtige Quelle chronischer Entzündung dämpfen und gleichzeitig einem anderen Weg erlauben, die antiviralen Interferonsignale zu erzeugen. Wenn sich diese Strategie in weiteren Studien als sicher erweist, könnte sie gezielte Behandlungen nicht nur für seltene erbliche Fieber‑Syndrome wie FCAS eröffnen, sondern auch für weiter gefasste Zustände des „Inflammaging“, in denen anhaltende, niedriggradige Entzündung Krankheiten antreibt.
Zitation: Lackner, A., Picucci, S.I., Jiang, W. et al. TH5487 specifically targets NLRP3 in FCAS patients resistant to MCC950. Commun Biol 9, 528 (2026). https://doi.org/10.1038/s42003-026-10008-2
Schlüsselwörter: NLRP3-Inflammasom, oxidierte mitochondriale DNA, TH5487, cGAS-STING-Weg, autoinflammatorische Erkrankung