Clear Sky Science · en
TH5487 specifically targets NLRP3 in FCAS patients resistant to MCC950
Why calming runaway inflammation matters
Many chronic diseases, from rare fevers to age-related disorders, are driven by an immune alarm system that refuses to switch off. This study explores new drug candidates that can quiet one of the body’s most powerful inflammatory switches, called NLRP3, without shutting down our defenses altogether. By rethinking existing molecules originally aimed at DNA repair, the authors uncover a fresh way to rein in harmful inflammation and even redirect it into a more protective antiviral-style response.
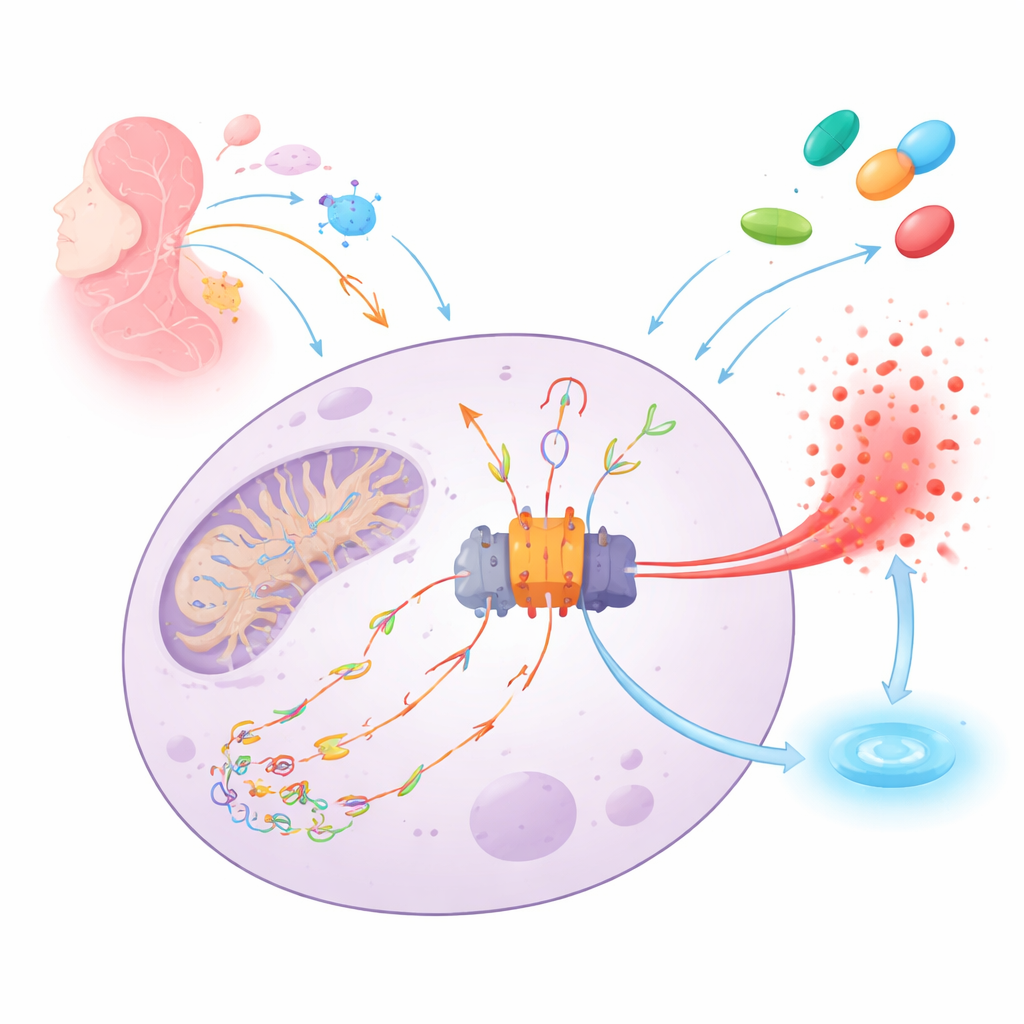
The body’s fire alarm and its spark
NLRP3 acts like a molecular smoke detector inside immune cells. When it senses danger—ranging from infections to tissue damage—it assembles a large protein machine (the inflammasome) that triggers the release of the potent alarm signal IL‑1β and can rupture the cell to warn its neighbors. A key spark for this alarm is damaged mitochondrial DNA that has been nicked and oxidized by reactive oxygen species. These damaged DNA fragments can leak out of mitochondria, float through the cell, bind NLRP3, and drive inflammasome assembly and inflammatory cell death.
Old DNA repair drugs, new anti-inflammatory roles
The authors examined small molecules first designed to target a DNA repair enzyme called hOGG1, which removes oxidized bases from DNA. Surprisingly, these drugs—TH5487, SU0268, and an activator named TH10785—also interact with NLRP3. In human blood immune cells and model cell lines, low micromolar doses of TH5487 and SU0268 sharply reduced IL‑1β release and the activation of caspase‑1, the enzyme that matures this cytokine. Detailed protein pull‑down and imaging experiments showed that the drugs prevent NLRP3 from assembling its full inflammasome complex: they block the recruitment of key partners (ASC, NEK7, and pro‑caspase‑1) and reduce the formation of the bright “specks” that mark an active inflammasome.
Cutting the link between damaged DNA and the inflammasome
Because mitochondrial damage is a major driver of NLRP3 activity, the team tested whether the drugs could block inflammasome activation triggered by mitochondrial stress. A compound that disrupts the cell’s energy factories and boosts reactive oxygen species normally sends oxidized mitochondrial DNA toward NLRP3 and strongly raises IL‑1β levels. TH5487 and SU0268 not only cut IL‑1β release under these conditions, they also reduced the amount of NLRP3 found at mitochondria. Cryo‑electron microscopy confirmed that NLRP3 can form a large ring‑like complex that physically binds mitochondrial DNA, and computer modeling suggested that the protein’s pyrin domain recognizes oxidized single‑stranded DNA via a positively charged surface patch. The drugs interfere with this DNA‑sensing step, severing a crucial link between damaged DNA and runaway inflammation.
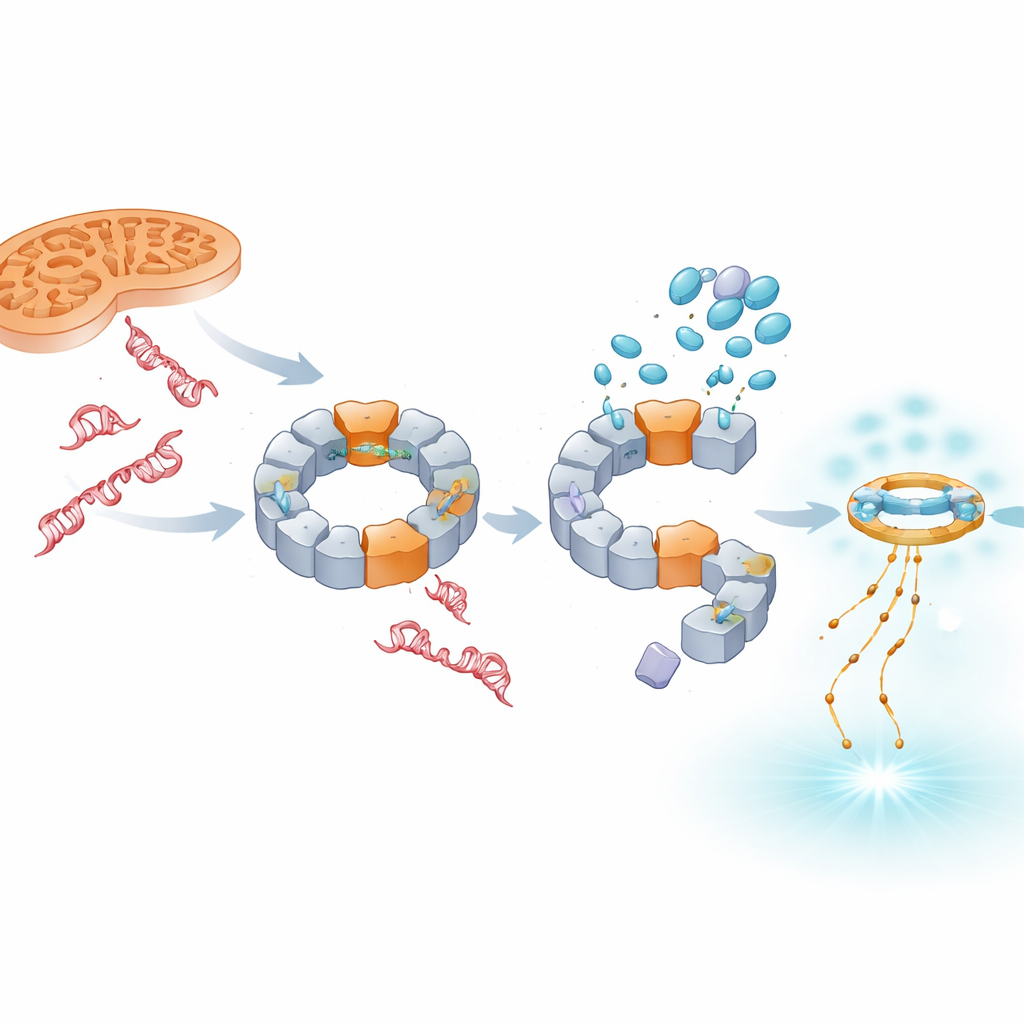
Shifting the signal toward antiviral-style defenses
Blocking NLRP3 does not make damaged DNA disappear; instead, more of it, especially from the nucleus, accumulates in the cell’s fluid. The study shows that this excess DNA is picked up by another sensor system, cGAS–STING, which typically responds to viral genomes. When TH5487, SU0268, or TH10785 are present, levels of activated STING and the antiviral messenger IFN‑β rise in both model cells and primary human blood cells. Importantly, this boost occurs even when NLRP3 is genetically removed, indicating that cGAS–STING acts as an independent backup alarm. Thus, these compounds quiet IL‑1β‑driven inflammation while tilting the immune response toward interferon‑based signaling that may be more protective in chronic disease.
Promise for difficult-to-treat inherited inflammation
A particularly striking test involved Familial Cold Autoinflammatory Syndrome (FCAS), a rare disorder where mutations in NLRP3 make it hyperactive and resistant to a leading experimental inhibitor, MCC950. In immune cells from mice and human patients carrying these mutations, MCC950 failed to curb IL‑1β release. TH5487 and SU0268, however, significantly reduced this overproduction at doses that spared cell viability. This suggests that targeting the oxidized DNA–sensing surface of NLRP3, rather than its more traditional enzymatic core, may bypass resistance and offer new treatment options for patients whose disease currently has limited targeted therapies.
What these findings mean for future therapies
To a non-specialist, the core message is that the researchers have found a new “off switch” for a powerful inflammatory machine inside our cells. By repurposing DNA repair drugs to block how NLRP3 senses damaged mitochondrial and nuclear DNA, they can dampen a major source of chronic inflammation while allowing another pathway to take over and produce antiviral interferon signals. If this strategy proves safe in further studies, it could open up targeted treatments not only for rare inherited fever syndromes like FCAS, but also for broader conditions of “inflammaging” in which persistent, low‑grade inflammation fuels disease.
Citation: Lackner, A., Picucci, S.I., Jiang, W. et al. TH5487 specifically targets NLRP3 in FCAS patients resistant to MCC950. Commun Biol 9, 528 (2026). https://doi.org/10.1038/s42003-026-10008-2
Keywords: NLRP3 inflammasome, oxidized mitochondrial DNA, TH5487, cGAS-STING pathway, autoinflammatory disease