Clear Sky Science · pl
TH5487 celuje specyficznie w NLRP3 u pacjentów z FCAS opornych na MCC950
Dlaczego ważne jest uspokojenie wymykającego się zapalenia
Wiele chorób przewlekłych, od rzadkich gorączek po schorzenia związane z wiekiem, napędzanych jest przez układ alarmowy odporności, który odmawia wyłączenia. Badanie to bada nowe kandydaty na leki, które mogą uciszyć jeden z najpotężniejszych zapalnych „wyłączników” w organizmie, zwany NLRP3, bez całkowitego wyłączania naszej obrony. Przeanalizowawszy na nowo istniejące cząsteczki początkowo skierowane przeciwko naprawie DNA, autorzy odkrywają nowy sposób powstrzymywania szkodliwego zapalenia, a nawet przekształcania go w bardziej ochronną odpowiedź w stylu przeciwwirusowym.
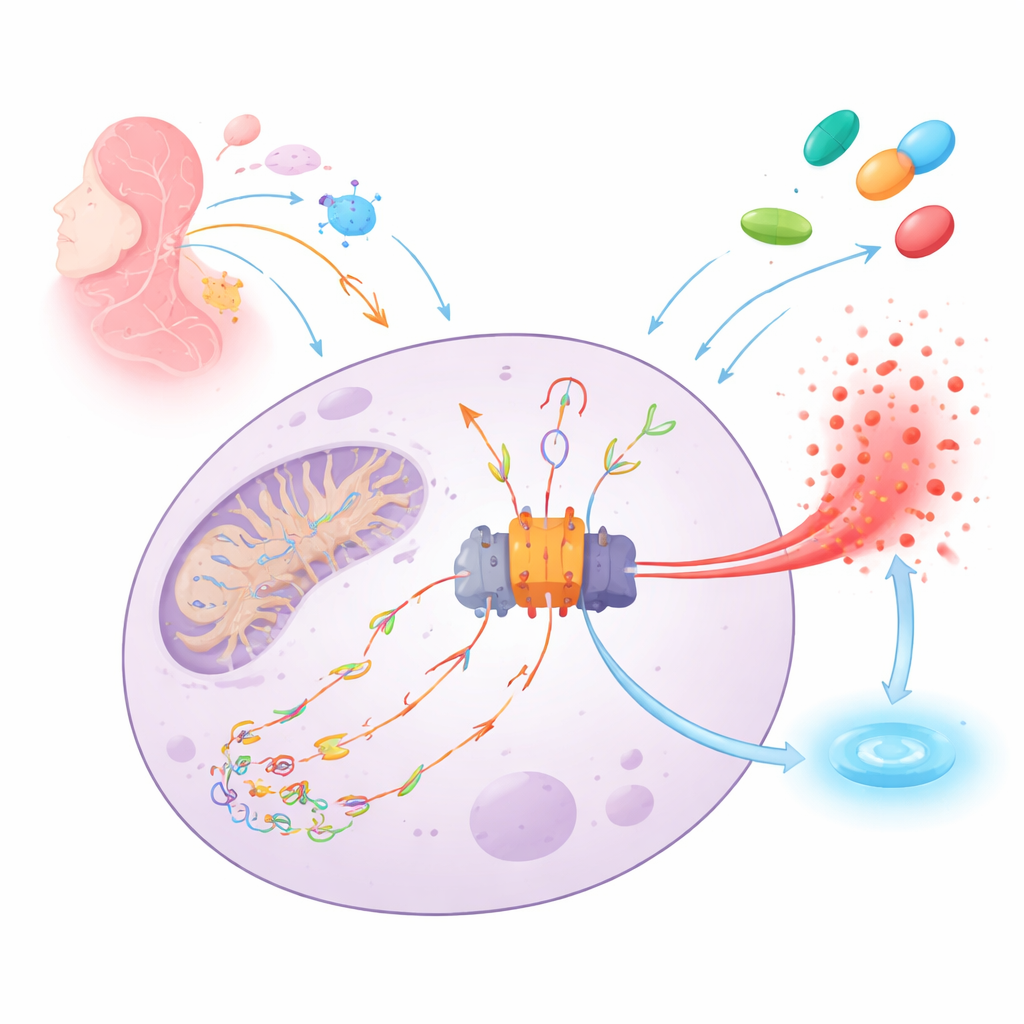
System alarmowy ciała i jego iskra
NLRP3 działa jak molekularny czujnik dymu wewnątrz komórek odpornościowych. Gdy wykryje zagrożenie — od infekcji po uszkodzenia tkanek — składa się w duży kompleks białkowy (inflammasom), który wyzwala uwalnianie silnego sygnału alarmowego IL‑1β i może doprowadzić do pęknięcia komórki, by ostrzec sąsiadów. Kluczową iskrą dla tego alarmu jest uszkodzone DNA mitochondrialne, które zostało nadłamane i zoksydowane przez reaktywne formy tlenu. Te uszkodzone fragmenty DNA mogą wydostawać się z mitochondriów, przemieszczać się przez komórkę, wiązać NLRP3 i wymuszać składanie inflammasomu oraz zapalną śmierć komórki.
Stare leki naprawiające DNA, nowe role przeciwzapalne
Autorzy zbadali małe cząsteczki pierwotnie zaprojektowane do celowania w enzym naprawy DNA o nazwie hOGG1, usuwający zoksydowane zasady z DNA. Ku zaskoczeniu, te leki — TH5487, SU0268 oraz aktywator TH10785 — wchodzą też w interakcje z NLRP3. W ludzkich komórkach krwi i liniach modelowych niskie mikromolowe dawki TH5487 i SU0268 znacznie zmniejszały uwalnianie IL‑1β i aktywację kaspazy‑1, enzymu dojrzewiającego ten cytokin. Szczegółowe eksperymenty z wyłapywaniem białek i obrazowaniem wykazały, że leki zapobiegają złożeniu pełnego kompleksu inflammasomu NLRP3: blokują rekrutację kluczowych partnerów (ASC, NEK7 i pro‑kaspazy‑1) oraz redukują powstawanie jasnych „plamek” oznaczających aktywny inflammasom.
Przecięcie łącza między uszkodzonym DNA a inflammasomem
Ponieważ uszkodzenie mitochondriów jest głównym czynnikiem napędzającym aktywność NLRP3, zespół sprawdził, czy leki mogą zablokować aktywację inflammasomu wywołaną stresem mitochondrialnym. Związek zaburzający działanie „elektrowni” komórkowych i zwiększający formy tlenu zwykle kieruje zoksydowane DNA mitochondrialne do NLRP3 i silnie podnosi poziomy IL‑1β. TH5487 i SU0268 nie tylko obniżały uwalnianie IL‑1β w tych warunkach, lecz także zmniejszały ilość NLRP3 obecnego przy mitochondriach. Krio‑mikroskopia elektronowa potwierdziła, że NLRP3 może tworzyć duży pierścieniowaty kompleks, który fizycznie wiąże DNA mitochondrialne, a modelowanie komputerowe zasugerowało, że domena pyrin białka rozpoznaje zoksydowane jednoniciowe DNA przez dodatnio naładowaną powierzchnię. Leki zakłócają ten etap rozpoznawania DNA, przerywając kluczowe połączenie między uszkodzonym DNA a wymykającym się zapaleniem.
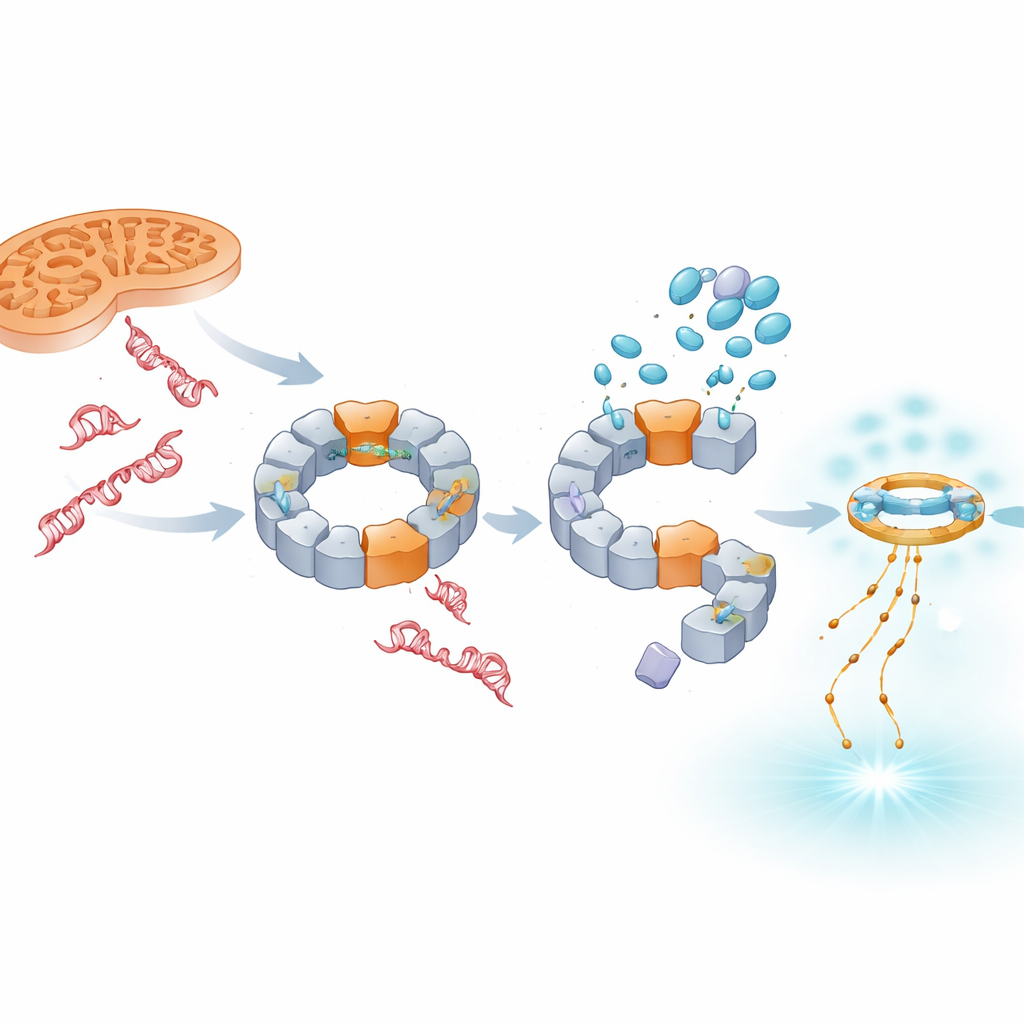
Przesunięcie sygnału w stronę obrony w stylu przeciwwirusowym
Zablokowanie NLRP3 nie usuwa uszkodzonego DNA; zamiast tego więcej takiego DNA, szczególnie pochodzącego z jądra, gromadzi się w płynie komórkowym. Badanie wykazuje, że nadmiar tego DNA jest wychwytywany przez inny układ czujników, cGAS–STING, który zwykle reaguje na genomy wirusowe. W obecności TH5487, SU0268 lub TH10785 wzrastają poziomy aktywowanego STING i przeciwwirusowego komunikatu IFN‑β zarówno w komórkach modelowych, jak i w pierwotnych komórkach krwi ludzkiej. Co istotne, to wzmocnienie występuje nawet gdy NLRP3 jest genetycznie usunięty, co wskazuje, że cGAS–STING działa jako niezależny alarm zastępczy. W ten sposób związki te tłumią zapalenie napędzane przez IL‑1β, jednocześnie przechylając odpowiedź immunologiczną w stronę sygnalizacji interferonowej, która może być bardziej ochronna w chorobach przewlekłych.
Obietnica dla trudnych do leczenia zapaleń dziedzicznych
Szczególnie przekonujący test dotyczył Rodzinnego Zimowego Zespołu Autoinflamacyjnego (FCAS), rzadkiego schorzenia, w którym mutacje w NLRP3 czynią go nadaktywnego i opornego na wiodący eksperymentalny inhibitor MCC950. W komórkach odpornościowych myszy i pacjentów ludzkich niosących te mutacje MCC950 nie zdołał powstrzymać uwalniania IL‑1β. TH5487 i SU0268 natomiast znacząco zmniejszały tę nadprodukcję w dawkach oszczędzających żywotność komórek. Sugeruje to, że celowanie w powierzchnię NLRP3 rozpoznającą zoksydowane DNA — zamiast jego tradycyjnego enzymatycznego rdzenia — może obejść oporność i zaoferować nowe opcje leczenia dla pacjentów, których choroba obecnie ma ograniczone terapie ukierunkowane.
Co te odkrycia znaczą dla przyszłych terapii
Dla osoby niebędącej specjalistą główny wniosek jest taki, że badacze znaleźli nowy „wyłącznik” dla potężnej zapalnej maszyny wewnątrz naszych komórek. Poprzez repurposing leków naprawiających DNA, by blokowały rozpoznawanie uszkodzonego DNA mitochondrialnego i jądrowego przez NLRP3, można stłumić ważne źródło przewlekłego zapalenia, pozwalając jednocześnie innemu szlakowi przejąć funkcję i wytwarzać przeciwwirusowe sygnały interferonowe. Jeśli ta strategia okaże się bezpieczna w dalszych badaniach, może otworzyć drogę do ukierunkowanych terapii nie tylko dla rzadkich dziedzicznych zespołów gorączkowych jak FCAS, ale także dla szerszych stanów „inflammaging”, w których utrzymujące się, niskiego stopnia zapalenie napędza choroby.
Cytowanie: Lackner, A., Picucci, S.I., Jiang, W. et al. TH5487 specifically targets NLRP3 in FCAS patients resistant to MCC950. Commun Biol 9, 528 (2026). https://doi.org/10.1038/s42003-026-10008-2
Słowa kluczowe: inflammasom NLRP3, zoksydowane DNA mitochondrialne, TH5487, szlak cGAS-STING, choroba autoinflamacyjna