Clear Sky Science · fr
Le TH5487 cible spécifiquement NLRP3 chez des patients FCAS résistants à MCC950
Pourquoi il est crucial d’apaiser une inflammation incontrôlée
De nombreuses maladies chroniques, des fièvres rares aux troubles liés à l’âge, sont alimentées par un système d’alarme immunitaire qui refuse de s’éteindre. Cette étude explore de nouveaux candidats médicaments capables d’endiguer l’un des interrupteurs inflammatoires les plus puissants du corps, appelé NLRP3, sans pour autant neutraliser nos défenses. En repensant des molécules existantes initialement destinées à la réparation de l’ADN, les auteurs dévoilent une nouvelle façon de maîtriser l’inflammation délétère et même de la rediriger vers une réponse plus protectrice de type antiviral.
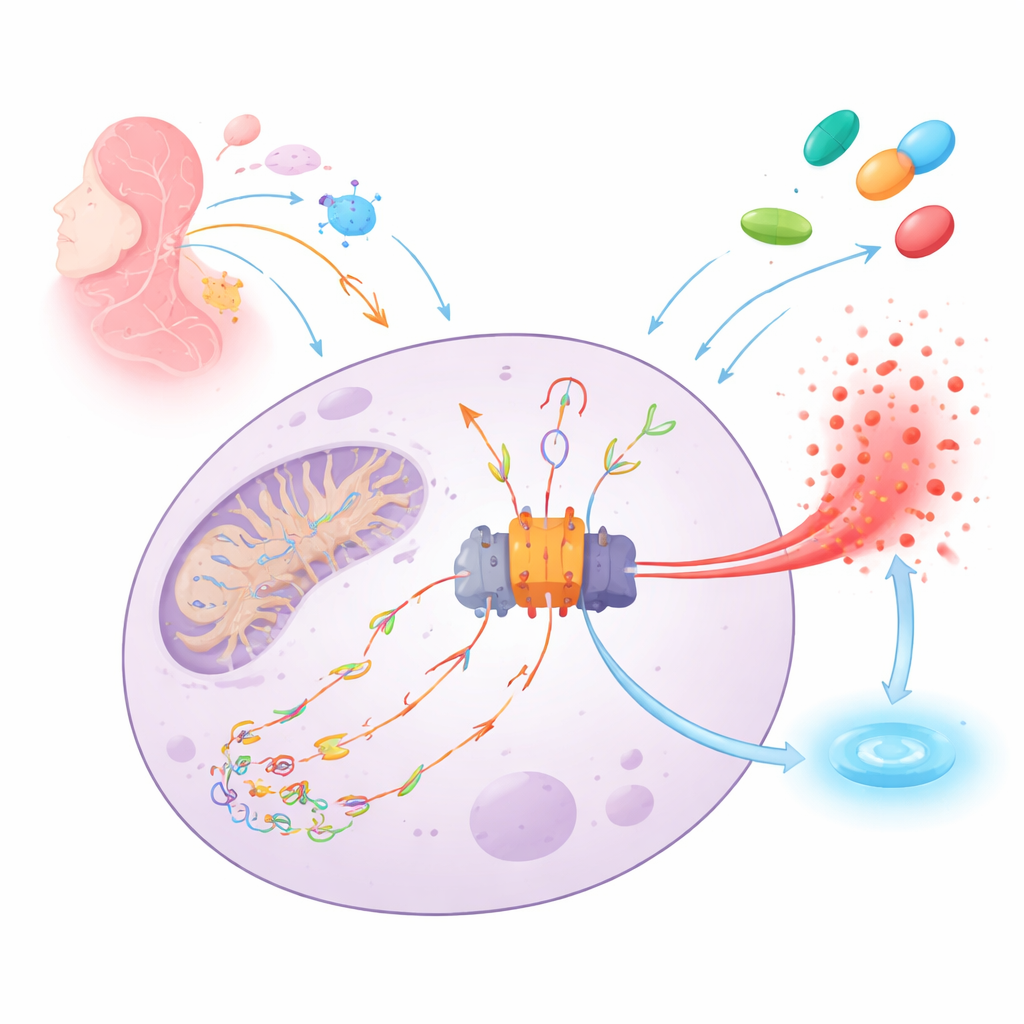
L’alarme incendie du corps et son étincelle
NLRP3 fonctionne comme un détecteur de fumée moléculaire au sein des cellules immunitaires. Lorsqu’il perçoit un danger — qu’il s’agisse d’infections ou de lésions tissulaires — il assemble une vaste machinerie protéique (l’inflammasome) qui déclenche la libération du puissant signal d’alarme IL‑1β et peut provoquer la rupture cellulaire pour avertir les voisines. Une étincelle clé pour cette alarme est l’ADN mitochondrial endommagé, entaillé et oxydé par des espèces réactives de l’oxygène. Ces fragments d’ADN altérés peuvent fuir des mitochondries, circuler dans la cellule, se lier à NLRP3 et favoriser l’assemblage de l’inflammasome ainsi que la mort cellulaire inflammatoire.
Anciens médicaments de réparation de l’ADN, nouveaux rôles anti-inflammatoires
Les auteurs ont examiné des petites molécules initialement conçues pour cibler une enzyme de réparation de l’ADN appelée hOGG1, qui élimine les bases oxydées de l’ADN. De manière surprenante, ces médicaments — TH5487, SU0268, et un activateur nommé TH10785 — interagissent aussi avec NLRP3. Dans des cellules immunes sanguines humaines et des lignées cellulaires modèles, de faibles doses micromolaires de TH5487 et SU0268 ont fortement réduit la libération d’IL‑1β et l’activation de la caspase‑1, l’enzyme qui mâture cette cytokine. Des expériences détaillées d’extraction de protéines et d’imagerie ont montré que ces composés empêchent NLRP3 d’assembler son complexe inflammasome complet : ils bloquent le recrutement de partenaires clés (ASC, NEK7 et pro‑caspase‑1) et réduisent la formation des « specks » lumineux qui marquent un inflammasome actif.
Couper le lien entre l’ADN endommagé et l’inflammasome
Parce que les dommages mitochondriaux sont un moteur majeur de l’activité de NLRP3, l’équipe a testé si les composés pouvaient bloquer l’activation de l’inflammasome déclenchée par le stress mitochondrial. Un composé qui perturbe les centrales énergétiques de la cellule et augmente les espèces réactives de l’oxygène envoie normalement de l’ADN mitochondrial oxydé vers NLRP3 et élève fortement les niveaux d’IL‑1β. TH5487 et SU0268 ont non seulement réduit la libération d’IL‑1β dans ces conditions, mais ont aussi diminué la quantité de NLRP3 retrouvée aux mitochondries. La cryo‑microscopie électronique a confirmé que NLRP3 peut former un grand complexe en forme d’anneau qui se lie physiquement à l’ADN mitochondrial, et la modélisation informatique suggère que le domaine pyrine de la protéine reconnaît l’ADN simple brin oxydé via une zone de surface chargée positivement. Les médicaments interfèrent avec cette étape de détection de l’ADN, rompant un lien crucial entre l’ADN endommagé et l’inflammation incontrôlée.
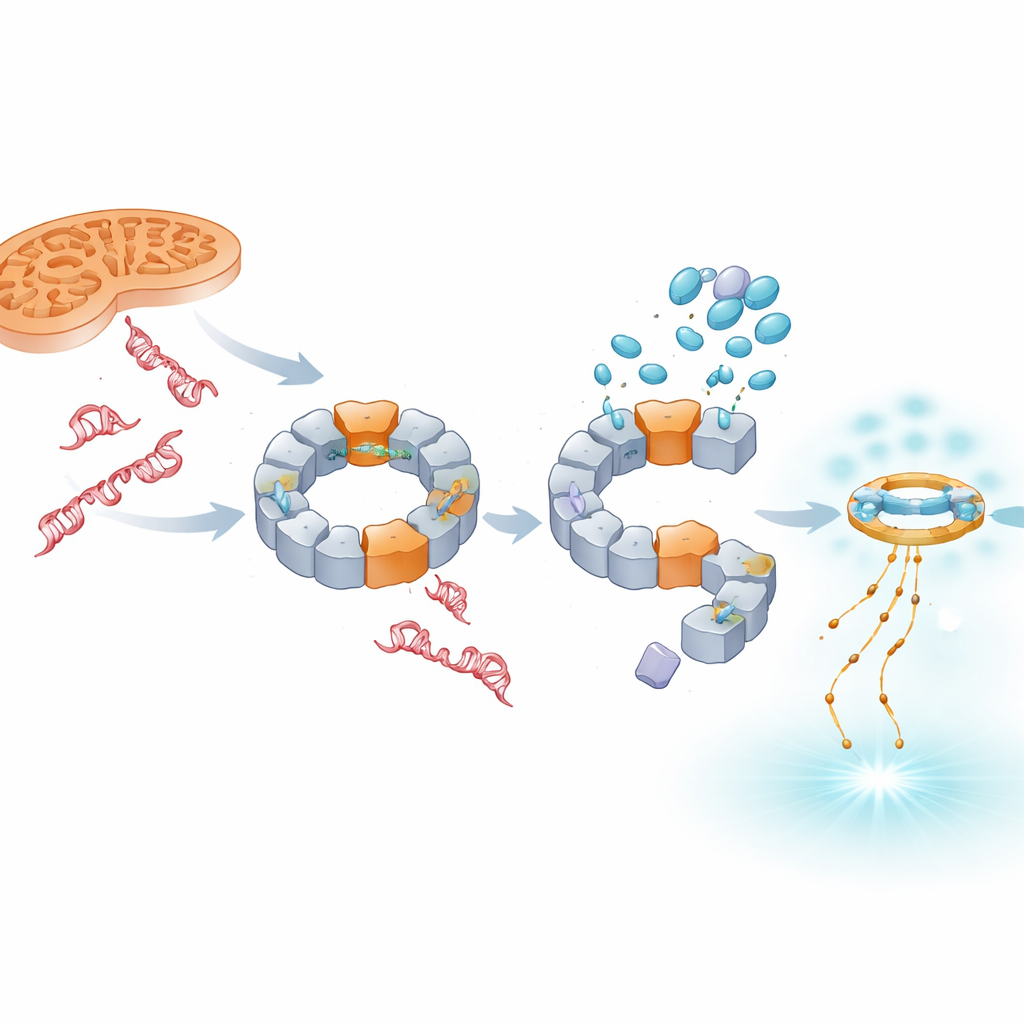
Faire basculer le signal vers des défenses de type antiviral
Bloquer NLRP3 ne fait pas disparaître l’ADN endommagé ; au contraire, une plus grande partie, notamment d’origine nucléaire, s’accumule dans le cytosol. L’étude montre que cet ADN en excès est capté par un autre système senseur, cGAS–STING, qui répond typiquement aux génomes viraux. En présence de TH5487, SU0268 ou TH10785, les niveaux de STING activé et du messager antiviral IFN‑β augmentent dans les cellules modèles comme dans des cellules sanguines humaines primaires. Il est important de noter que cette hausse se produit même lorsque NLRP3 est supprimé génétiquement, ce qui indique que cGAS–STING agit comme une alarme de secours indépendante. Ainsi, ces composés calment l’inflammation pilotée par l’IL‑1β tout en inclinant la réponse immunitaire vers une signalisation à base d’interférons qui peut être plus protectrice en cas de maladie chronique.
Promesse pour des inflammations héréditaires difficiles à traiter
Un испытание particulièrement révélateur a porté sur le syndrome autoinflammatoire familial au froid (FCAS), un trouble rare où des mutations de NLRP3 le rendent hyperactif et résistant à un inhibiteur expérimental majeur, MCC950. Dans des cellules immunes de souris et de patients humains portant ces mutations, MCC950 n’a pas réussi à réduire la libération d’IL‑1β. TH5487 et SU0268 ont cependant diminué de manière significative cette surproduction à des doses épargnant la viabilité cellulaire. Cela suggère que cibler la surface de détection de l’ADN oxydé de NLRP3, plutôt que son noyau enzymatique plus traditionnel, peut contourner la résistance et offrir de nouvelles options thérapeutiques pour des patients dont la maladie dispose aujourd’hui de peu de traitements ciblés.
Ce que ces découvertes signifient pour les thérapies futures
Pour un non‑spécialiste, le message central est que les chercheurs ont identifié un nouvel « interrupteur off » pour une puissante machinerie inflammatoire à l’intérieur de nos cellules. En réaffectant des médicaments de réparation de l’ADN pour bloquer la manière dont NLRP3 détecte l’ADN mitochondrial et nucléaire endommagé, ils peuvent atténuer une source majeure d’inflammation chronique tout en laissant une autre voie prendre le relais et produire des signaux interféronaux antiviraux. Si cette stratégie s’avère sûre dans des études ultérieures, elle pourrait ouvrir des traitements ciblés non seulement pour des syndromes fébriles héréditaires rares comme le FCAS, mais aussi pour des affections plus larges d’« inflammaging » où une inflammation persistante et de bas grade alimente la maladie.
Citation: Lackner, A., Picucci, S.I., Jiang, W. et al. TH5487 specifically targets NLRP3 in FCAS patients resistant to MCC950. Commun Biol 9, 528 (2026). https://doi.org/10.1038/s42003-026-10008-2
Mots-clés: inflammasome NLRP3, ADN mitochondrial oxydé, TH5487, voie cGAS-STING, maladie autoinflammatoire