Clear Sky Science · sv
En datorbaserad regelstyrd modell för reglering av MAPK/ERK-systemet
Varför små cellulära brytare spelar roll
Varje sekund måste våra celler avgöra om de ska växa, dela sig, röra sig eller självdestrueras. En central beslutsfattare är en kedja av proteiner kallad MAPK/ERK-vägen. Den är också ett av de mest frekvent störda systemen vid cancer. Fastän läroböcker ofta visar denna väg som ett enkelt relä, är den i själva verket ett tätt nätverk av interagerande delar. Denna studie bygger en detaljerad datorbaserad modell av det nätverket för att ställa en praktisk fråga: hur kan en enda väg samtidigt ge milda, graderade svar och skarpa, allt-eller-inget-beslut, och hur samarbetar eller konkurrerar nära besläktade proteiner för att styra cellödet?
En trafikerad motorväg inne i cellen
MAPK/ERK-vägen börjar vid cellens yta, där tillväxtfaktorer binder receptorer och rekryterar ett hjälpprotein kallat SOS. SOS aktiverar RAS, en liten molekylär strömbrytare förankrad i cellmembranets insida. Aktiv RAS rekryterar sedan en familj enzymer som kallas RAF, vilka aktiverar MEK, som i sin tur aktiverar ERK. Aktiverat ERK reglerar hundratals mål i hela cellen och påverkar om cellen prolifererar, differentierar, migrerar eller genomgår programmerad celldöd. Istället för att vara en enkel rak linje är denna bana full av återkopplingsslingor, förgreningar och parallella varianter av samma proteiner (isoformer), vilket gör dess beteende starkt kontextberoende.
Från enkel kaskad till regelbaserat nätverk
För att fånga denna komplexitet använder författarna regelbaserad modellering, en metod som kodar interaktionsmönster istället för att lista varje möjlig molekylär tillstånd för sig. Deras modell inkluderar två varianter av MEK (MEK1 och MEK2) och tre varianter av RAF (BRAF, CRAF och ARAF), liksom regulatoriska partner som 14-3-3-proteiner. Den bygger också in en förstärkande återkoppling (där aktiv RAS stärker sin egen aktivator SOS) och tre hämmande återkopplingar (där aktiv ERK dämpar SOS, RAF och MEK). Eftersom varje protein kan modifieras och binda på många olika sätt skulle en traditionell modell explodera i storlek; den regelbaserade metoden håller modellen hanterbar samtidigt som den representerar mer än 600 distinkta molekylära arter och tusentals reaktioner.
Att förklara graderade och brytarliknande signaler
Experiment har visat ett förbryllande mönster: på nivån RAS och RAF ökar aktiviteten gradvis när dosen av tillväxtfaktor stiger, men i slutet av kaskaden slår ERK ofta på i en nästan allt-eller-inget-maniér. Modellen förklarar detta genom att föreslå att RAS vid låga doser aktiveras endast i små fläckar på cellmembranet. När dosen ökar övergår fler delar av membranet till det aktiva tillståndet. Lokalt är RAS-brytaren bistabil—den hoppar skarpt från av till på—men eftersom endast en fraktion av membranet är aktivt framstår genomsnittlig RAS- och RAF-aktivitet över hela cellen som jämn. Nedströms fungerar den tvåstegsaktivering som MEK och ERK genomgår som en kraftfull förstärkare, så att även måttlig aktivitet uppströms ger nästan fullständig ERK-aktivering, vilket ger ett skarpt, brytarlikt svar. Negativ återkoppling från ERK till SOS omvandlar sedan stadig stimulering till rytmiska pulser av ERK-aktivitet, vars period beror på tillväxtfaktorns styrka och återkopplingens intensitet.
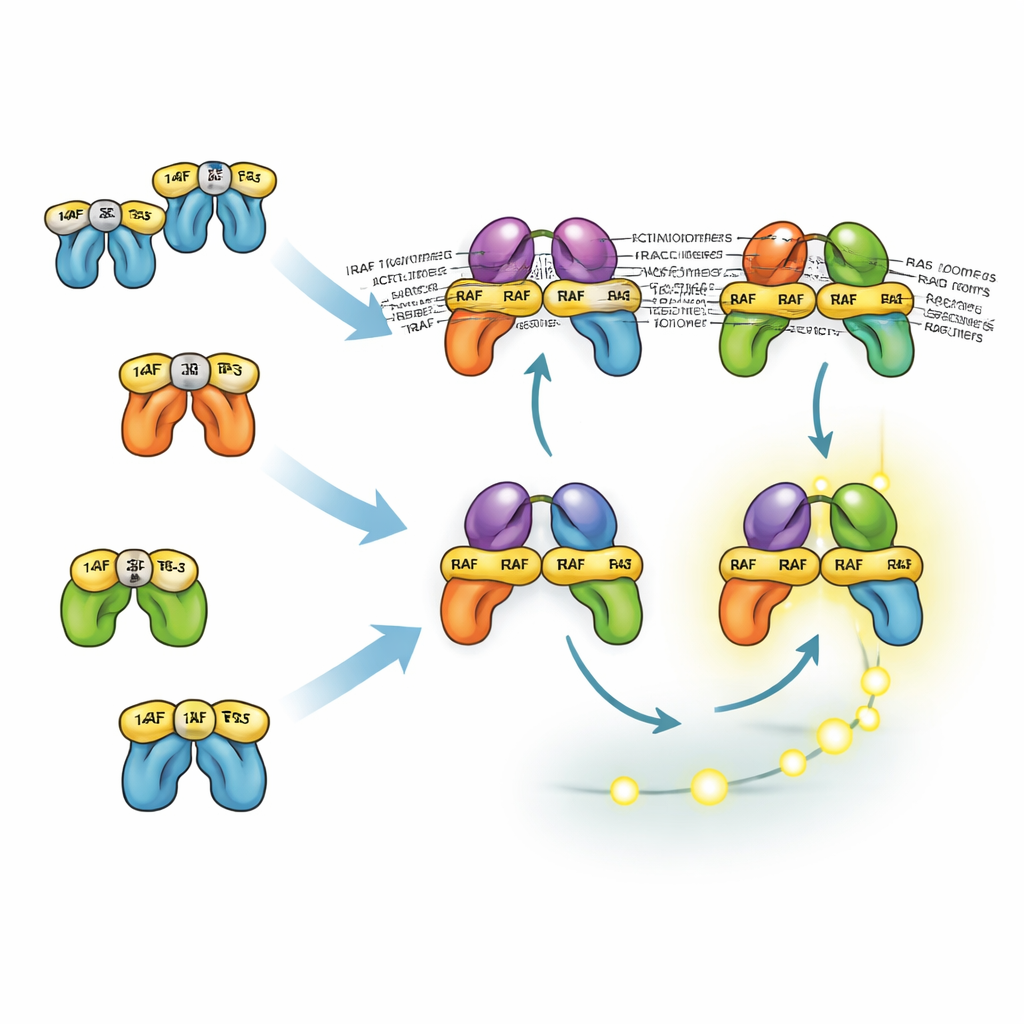
Hur liknande proteiner delar och fördelar uppgifter
De tre RAF-isoformerna beter sig olika på grund av hur de interagerar med 14-3-3-proteiner, som både kan låsa RAF-molekyler i en inaktiv form och stabilisera aktiva RAF-par. BRAF:s huvudsakliga dockningsplats för 14-3-3 ligger i dess svans, vilket gör att den kan förbli associerad med 14-3-3 samtidigt som den binder RAS och bildar dimerer med CRAF eller ARAF. Dessa blandade dimerer är särskilt stabila och effektiva vid att aktivera MEK. Däremot förlitar sig CRAF och ARAF mer på bindning i huvudänden till 14-3-3, vilket blockerar deras rekrytering till RAS. Som en följd är dimerer utan BRAF mindre stabila och oftast mindre vanliga. Modellen visar ändå att celler utan BRAF kan generera starka ERK-signaler eftersom uppströms positiv återkoppling och nedströms förstärkning kompenserar. Detta hjälper till att förklara varför läkemedel som riktar sig enbart mot BRAF ofta bara delvis undertrycker ERK och måste kombineras med MEK- eller ERK-hämmare.
Att balansera rörelse, överlevnad och död
Modellen spårar också RAF:s roller som går bortom att slå på ERK. När dimerer bryts upp under ERK-driven negativ återkoppling kan öppna former av CRAF binda partner som kontrollerar cellrörelse och överlevnad, såsom kinasen ROKα eller det pro-dödsfrämjande proteinet MST2. När tillväxtfaktornivåer och mängden 14-3-3 förändras skiftar balansen mellan BRAF-rika dimerer som starkt driver ERK, CRAF- och ARAF-baserade komplex som påverkar motilitet, och pooler som frigör MST2 för att främja celldöd. Eftersom dessa balanser svarar mjukt på stimulusstyrka medan ERK:s utsignal kan vara nästan allt-eller-inget, kan samma väg finjustera subtila beteendeaspekter och ändå fatta avgörande ödesval.
Vad detta betyder för förståelsen av celler
Genom att väva samman isoformspecifika interaktioner, lokal membranbrytning och inbäddade återkopplingsslingor visar modellen hur en molekylär väg kan fungera både som dimmer och som digital brytare. Variationer i mängderna av BRAF, CRAF, ARAF, MEK1, MEK2 och 14-3-3, som observerats i olika vävnader och tumörer, leder naturligt till olika dynamiska mönster av ERK-signalering, från pulser till bestående plattåer. Detta tyder på att celltypsspecifik koppling av samma kärnkomponenter kan återanvända MAPK/ERK-vägen för olika fysiologiska roller, och att effektiva terapier behöver beakta inte bara enskilda mål utan hela det regulatoriska sammanhang där dessa molekylära brytare verkar.
Citering: Kocieniewski, P., Lipniacki, T. A computational rule-based model of MAPK/ERK system regulation. Sci Rep 16, 14437 (2026). https://doi.org/10.1038/s41598-026-44353-3
Nyckelord: MAPK ERK-signalering, RAF-isoformer, 14-3-3-reglering, beräkningsmodell för väg, cellödesdynamik