Clear Sky Science · en
A computational rule-based model of MAPK/ERK system regulation
Why tiny cell switches matter
Every second, our cells must decide whether to grow, divide, move, or self-destruct. A central decision-maker is a chain of proteins called the MAPK/ERK pathway. It is also one of the most frequently disturbed systems in cancer. Although textbooks often show this pathway as a simple relay, it is in fact a dense web of interacting parts. This study builds a detailed computer model of that web to ask a practical question: how can one pathway simultaneously produce gentle, graded responses and sharp, all-or-nothing decisions, and how do closely related proteins cooperate or compete to control cell fate?
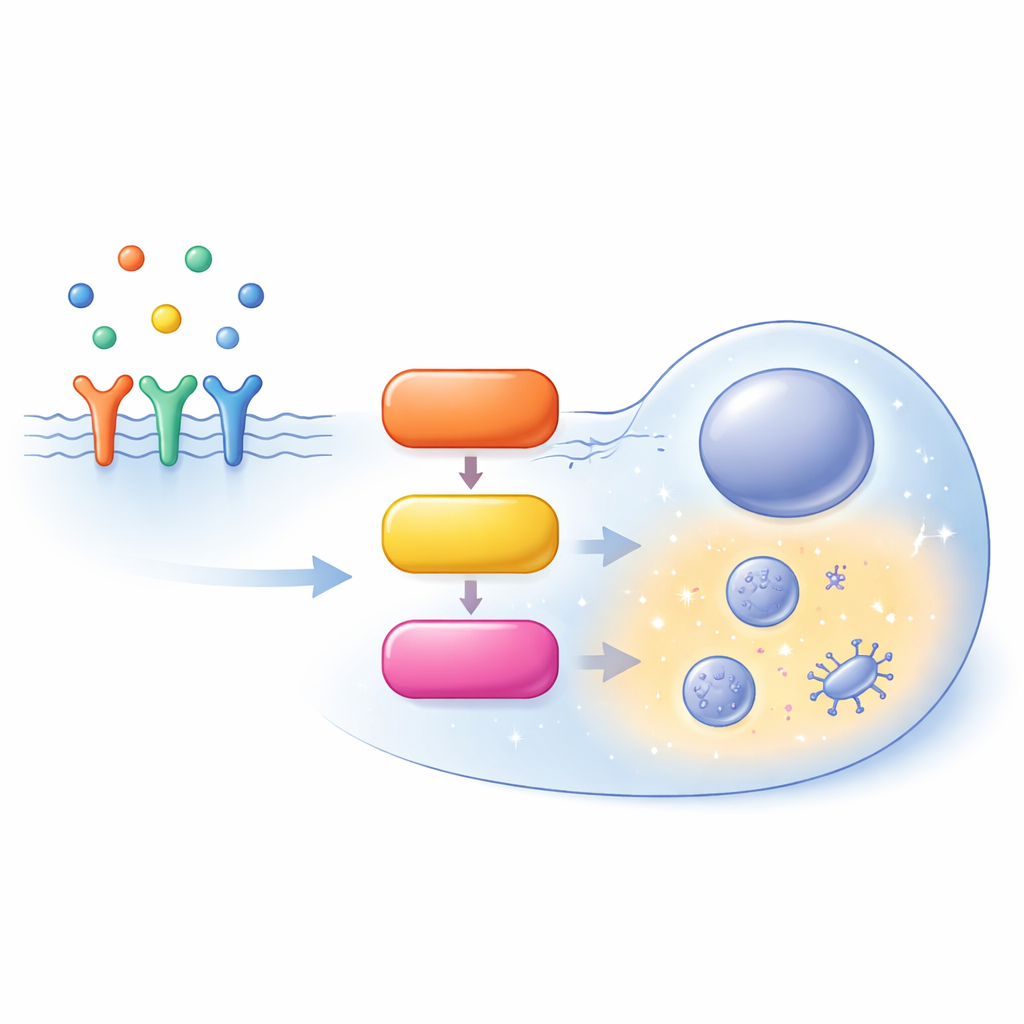
A busy highway inside the cell
The MAPK/ERK pathway starts at the cell surface, where growth factors bind to receptors and recruit a helper protein called SOS. SOS turns on RAS, a small molecular switch tethered to the inner face of the cell membrane. Active RAS then recruits a family of enzymes known as RAF, which activate MEK, which in turn activates ERK. Activated ERK goes on to regulate hundreds of targets throughout the cell, shaping whether the cell proliferates, differentiates, migrates, or undergoes programmed death. Rather than being a simple straight line, this route is full of feedback loops, branches, and parallel variants of the same proteins (isoforms), making its behavior highly context-dependent.
From simple cascade to rule-based network
To capture this complexity, the authors use rule-based modeling, a method that encodes interaction patterns instead of listing every possible molecular state one by one. Their model includes two versions of MEK (MEK1 and MEK2) and three versions of RAF (BRAF, CRAF, and ARAF), as well as regulatory partners such as 14-3-3 proteins. It also builds in one reinforcing feedback (where active RAS boosts its own activator SOS) and three braking feedbacks (where active ERK dampens SOS, RAF, and MEK). Because each protein can be modified and bound in many different ways, a traditional model would explode in size; the rule-based approach keeps it manageable while still representing more than 600 distinct molecular species and thousands of reactions.
Explaining graded and switch-like signals
Experiments have shown a puzzling pattern: at the level of RAS and RAF, activity increases gradually as the growth factor dose rises, but at the bottom of the cascade ERK often turns on in a near all-or-nothing fashion. The model explains this by proposing that, at low doses, RAS turns on only in small patches of the cell membrane. As the dose increases, more of the membrane flips into the active state. Locally, the RAS switch is bistable—it jumps sharply from off to on—but because only a fraction of the membrane is active, the average RAS and RAF activity across the whole cell appears smooth. Downstream, the dual-step activation of MEK and ERK acts like a powerful amplifier, so that even modest upstream activity produces almost complete ERK activation, yielding a sharp, switch-like response. Negative feedback from ERK to SOS then converts steady stimulation into rhythmic pulses of ERK activity, whose period depends on growth factor strength and feedback intensity.
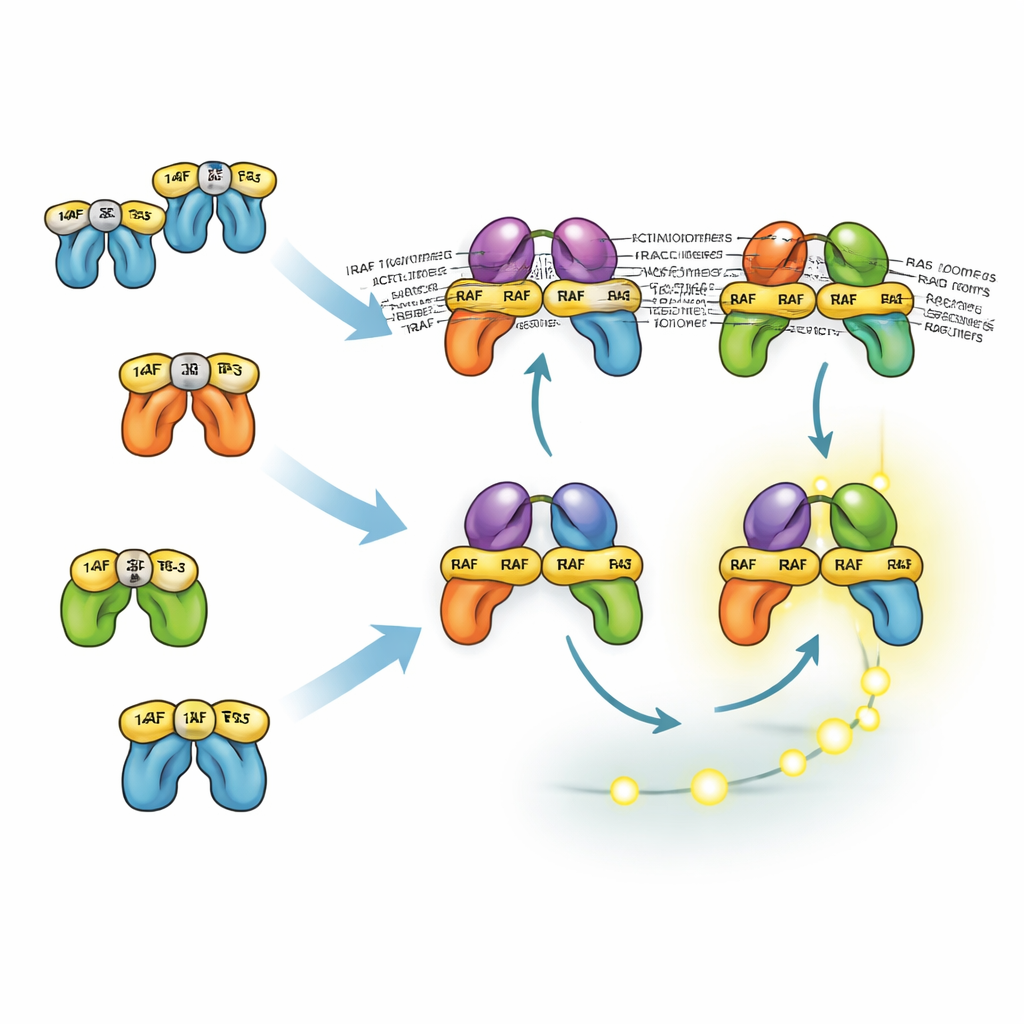
How look-alike proteins share and divide tasks
The three RAF isoforms behave differently because of how they interact with 14-3-3 proteins, which can both lock RAF molecules in an inactive shape and stabilize active pairs of RAF. BRAF’s main docking site for 14-3-3 lies at its tail, allowing it to stay associated with 14-3-3 while still binding RAS and forming dimers with CRAF or ARAF. These mixed dimers are especially stable and efficient at activating MEK. In contrast, CRAF and ARAF rely more on head-end binding to 14-3-3, which blocks their recruitment to RAS. As a result, dimers that lack BRAF are less stable and usually less abundant. Yet the model shows that, even without BRAF, cells can still generate strong ERK signals because upstream positive feedback and downstream amplification compensate. This helps explain why drugs that target BRAF alone often only partially suppress ERK and must be combined with MEK or ERK inhibitors.
Balancing movement, survival, and death
The model also tracks roles of RAF that go beyond switching on ERK. When dimers fall apart under ERK-driven negative feedback, open forms of CRAF can bind partners that control cell movement and survival, such as the kinase ROKα or the pro-death protein MST2. As growth factor levels and 14-3-3 abundance change, the balance shifts among BRAF-rich dimers that strongly drive ERK, CRAF- and ARAF-based complexes that modulate motility, and pools that free MST2 to promote cell death. Because these balances respond smoothly to stimulus strength while ERK’s output can be nearly all-or-none, the same pathway can tune subtle aspects of behavior and still make decisive fate choices.
What this means for understanding cells
By weaving together isoform-specific interactions, local membrane switching, and nested feedback loops, the model shows how one molecular pathway can act as both a dimmer and a digital switch. Variations in the amounts of BRAF, CRAF, ARAF, MEK1, MEK2, and 14-3-3, as observed across tissues and tumors, naturally lead to different dynamic patterns of ERK signaling, from pulses to sustained plateaus. This suggests that cell type–specific wiring of the same core components can repurpose the MAPK/ERK pathway for diverse physiological roles, and that effective therapies will need to consider not just single targets but the full regulatory context in which these molecular switches operate.
Citation: Kocieniewski, P., Lipniacki, T. A computational rule-based model of MAPK/ERK system regulation. Sci Rep 16, 14437 (2026). https://doi.org/10.1038/s41598-026-44353-3
Keywords: MAPK ERK signaling, RAF isoforms, 14-3-3 regulation, computational pathway model, cell fate dynamics