Clear Sky Science · sv
REDD1/DDIT4 motverkar endoplasmatiskt retikelstress‑inducerad apoptos genom att styra uttrycket av dödsreceptorn TRAILR2/DR5 i cancerceller
Varför stressade tumörceller spelar roll
Cancerceller växer inte under bekväma förhållanden. Inuti en tumör möter cellerna låg syretillgång, knappa näringsresurser och en ansamling av skadade proteiner. Under dessa påfrestningar dör vissa celler, men andra anpassar sig och blir svårare att slå ut med behandling. Den här studien undersöker hur kolonceller överlever en särskild typ av intern stress — problem i det endoplasmatiska retiklet, cellens proteinvikningsfabrik — och identifierar en molekylär ”broms” som förhindrar att dessa stressade celler självdö.
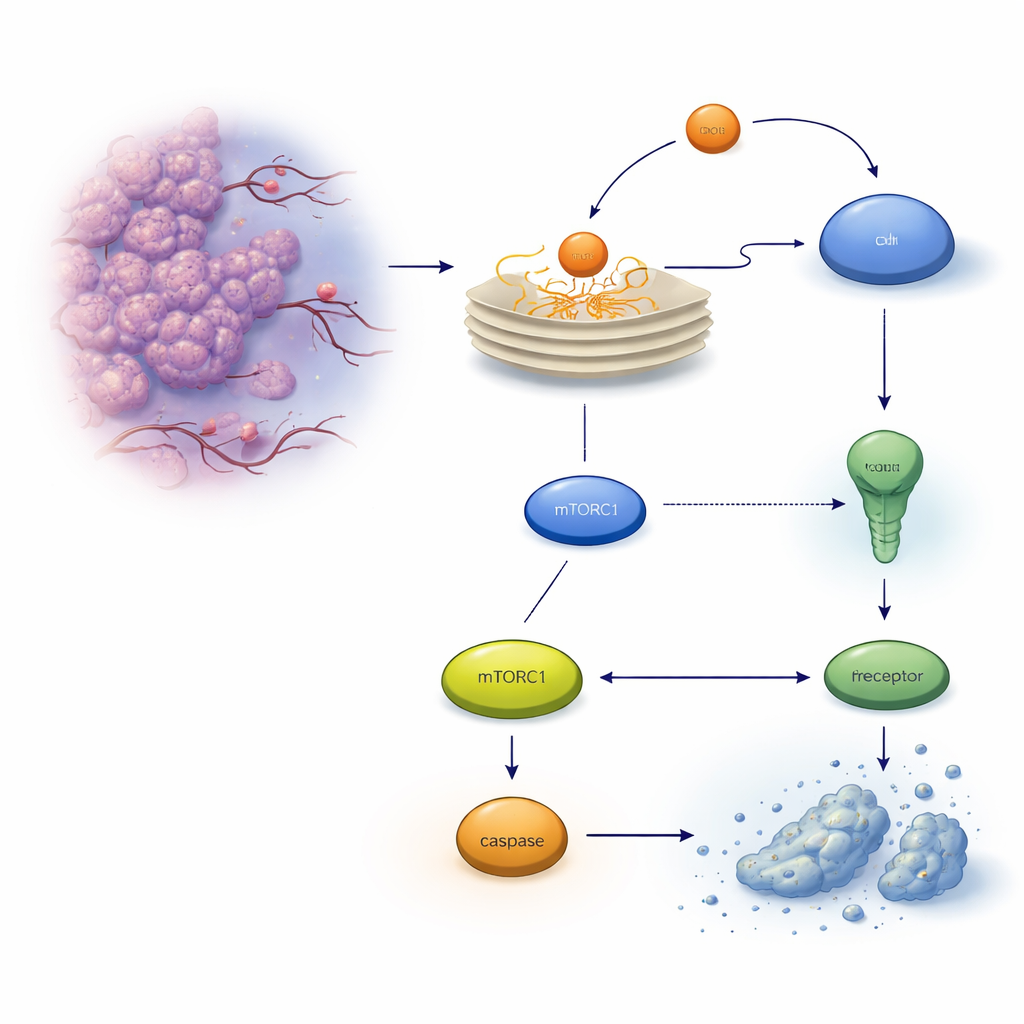
En dold säkerhetsventil inne i tumörceller
När proteiner inte viks korrekt ansamlas de i det endoplasmatiska retiklet (ER) och utlöser ett alarmsystem som kallas unfolded protein response. Inledningsvis försöker detta system återställa balansen, men om stressen är för stark eller varar för länge kan det övergå i ett självdestruktivt program kallat apoptos. I kolorektala cancerceller är en viktig effektor en dödsreceptor på cellytan som kallas TRAILR2/DR5, vilken kan aktivera en proteinnedbrytande enzymkaskad via caspas‑8 och i förlängningen döda cellen. Författarna fokuserade på ett annat stressinducerat protein, REDD1/DDIT4, redan känt för att hjälpa celler att anpassa sig till hårda förhållanden, och undersökte om det påverkar detta liv‑eller‑död‑val.
Hur ett stressprotein tippar balansen mot överlevnad
Forskarna utsatte humana koloncellinjer för läkemedel som orsakar ER‑stress och följde REDD1‑nivåerna över tid. De fann att ER‑stress snabbt ökade REDD1‑produktionen i en ordnad process styrd av en annan stressresponsiv faktor, ATF4. När REDD1 fanns närvarande dämpade det aktiviteten hos mTORC1, en central regulator av tillväxt och proteinsyntes. Att slå ner eller ta bort REDD1 med RNA‑interferens eller CRISPR‑genredigering gjorde cancercellerna mycket mer benägna att genomgå apoptos under ER‑stress, och denna ökade celldöd kunde blockeras med kaspasinhibitorer eller genom farmakologisk nedstängning av mTORC1, vilket visar att REDD1 normalt skyddar celler genom att begränsa denna tillväxtväg under stress.
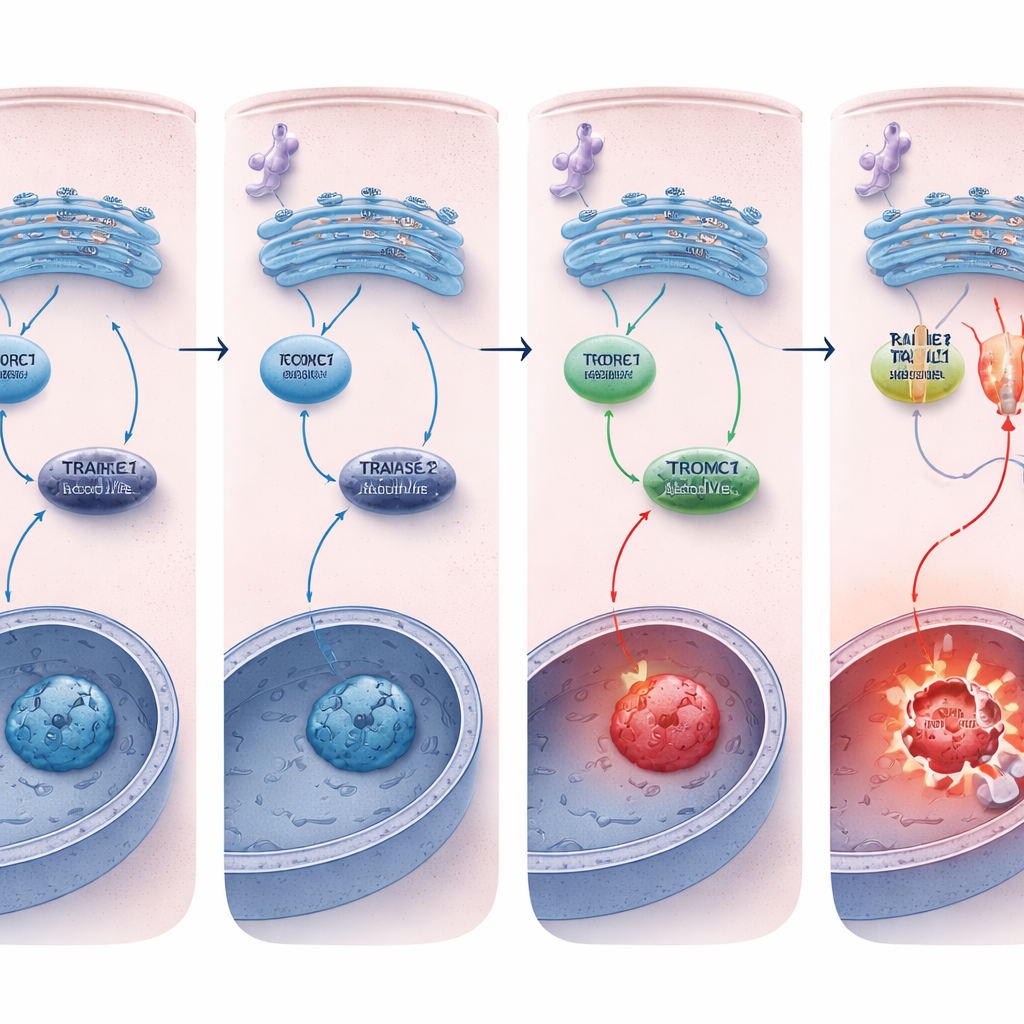
Att hålla dödsknappen nedtonad
Varför dog REDD1‑defekta celler lättare? Den avgörande skillnaden låg i dödsreceptorn TRAILR2/DR5. Vid kronisk ER‑stress aktiverade både normala och REDD1‑defekta celler de uppströms stressvägarna i liknande grad, men celler utan REDD1 visade en mycket större ökning av TRAILR2/DR5 på både RNA‑ och proteinnivå, tillsammans med kraftigare aktivering av caspas‑8. Tystnad av TRAILR2/DR5, men inte av en närliggande receptor, minskade tydligt apoptosen i dessa REDD1‑defekta celler, vilket bekräftar att den förhöjda dödsresponsen huvudsakligen drevs via denna receptor. I tredimensionella tumörsfäroider, som bättre efterliknar riktiga tumörer och naturligt ackumulerar stress, var REDD1‑nivåerna redan höga och mTORC1‑aktiviteten låg, och borttagning av REDD1 gjorde återigen dessa minitumörer betydligt mer sårbara för ER‑stress‑inducerad celldöd.
Roll för en transkriptionell broms
För att förstå hur REDD1 kontrollerar TRAILR2/DR5 använde teamet RNA‑sekvensering för att söka efter regulatorer vars nivåer förändrades när REDD1 saknades. De identifierade en transkriptionsfaktor kallad EVI‑1/MECOM, tidigare kopplad till tumörtillväxt och genrepression. I REDD1‑defekta celler sjönk nivåerna av EVI‑1/MECOM. Att minska EVI‑1/MECOM i annars normala celler gjorde dem mer känsliga för ER‑stress, ökade TRAILR2/DR5‑uttrycket och drev caspas‑8‑beroende apoptos, vilket efterliknade effekten av att förlora REDD1. Författarna fann också att corepressorer som CtBP1/2, kända samarbetspartner till EVI‑1/MECOM i vissa tumörer, ytterligare kunde justera TRAILR2/DR5‑nivåer och stresskänslighet på ett celltypsspecifikt sätt. Tillsammans tyder resultaten på att REDD1 hjälper till att upprätthålla EVI‑1/MECOM‑baserad repression av TRAILR2/DR5‑genen och därigenom fördröjer aktiveringen av dödsknappen under långvarig stress.
Vad detta betyder för framtida cancerbehandling
För en allmän publik är huvudbudskapet att kolorektala cancerceller har en intern mekanism som låter dem ”rida ut” svår proteinstress istället för att dö när de borde. Proteinet REDD1 fungerar som en säkerhetsventil: det dämpar en huvudväg för tillväxt och stöder ett genrepressionssystem som förhindrar att en potent dödsreceptor, TRAILR2/DR5, slås på för tidigt. När REDD1 eller dess partner EVI‑1/MECOM försvinner blir stressade cancerceller mycket mer benägna att självdö. Denna studie antyder att inriktning på REDD1 eller dess nedströms repressorer kan ta bort tumörernas skyddande buffert och göra cancerceller i fientliga, näringsfattiga delar av en tumör mer mottagliga för behandlingar som ökar ER‑stress och skjuter celler mot apoptos.
Citering: Mora-Molina, R., El Yousfi, Y., Hagenlocher, C. et al. REDD1/DDIT4 counteracts endoplasmic reticulum stress-induced apoptosis by controlling the expression of death receptor TRAILR2/DR5 in cancer cells. Cell Death Dis 17, 425 (2026). https://doi.org/10.1038/s41419-026-08648-7
Nyckelord: ER‑stress, kolorektal cancer, apoptos, TRAILR2/DR5, REDD1/DDIT4