Clear Sky Science · pl
REDD1/DDIT4 przeciwdziała apoptozie wywołanej stresem ER poprzez kontrolę ekspresji receptora śmierci TRAILR2/DR5 w komórkach nowotworowych
Dlaczego zestresowane komórki nowotworowe mają znaczenie
Komórki nowotworowe nie rosną w komfortowych warunkach. W obrębie guza komórki napotykają niskie stężenie tlenu, niedobór składników odżywczych i gromadzenie się uszkodzonych białek. Pod tym naciskiem część komórek ginie, ale inne adaptują się i stają się trudniejsze do zniszczenia terapią. Artykuł bada, jak komórki raka jelita grubego przetrwają jeden szczególny rodzaj wewnątrzkomórkowego stresu — zaburzenia w retikulum endoplazmatycznym, fabryce fałdowania białek — i identyfikuje molekularny „hamulec”, który zapobiega samozniszczeniu tych zestresowanych komórek.
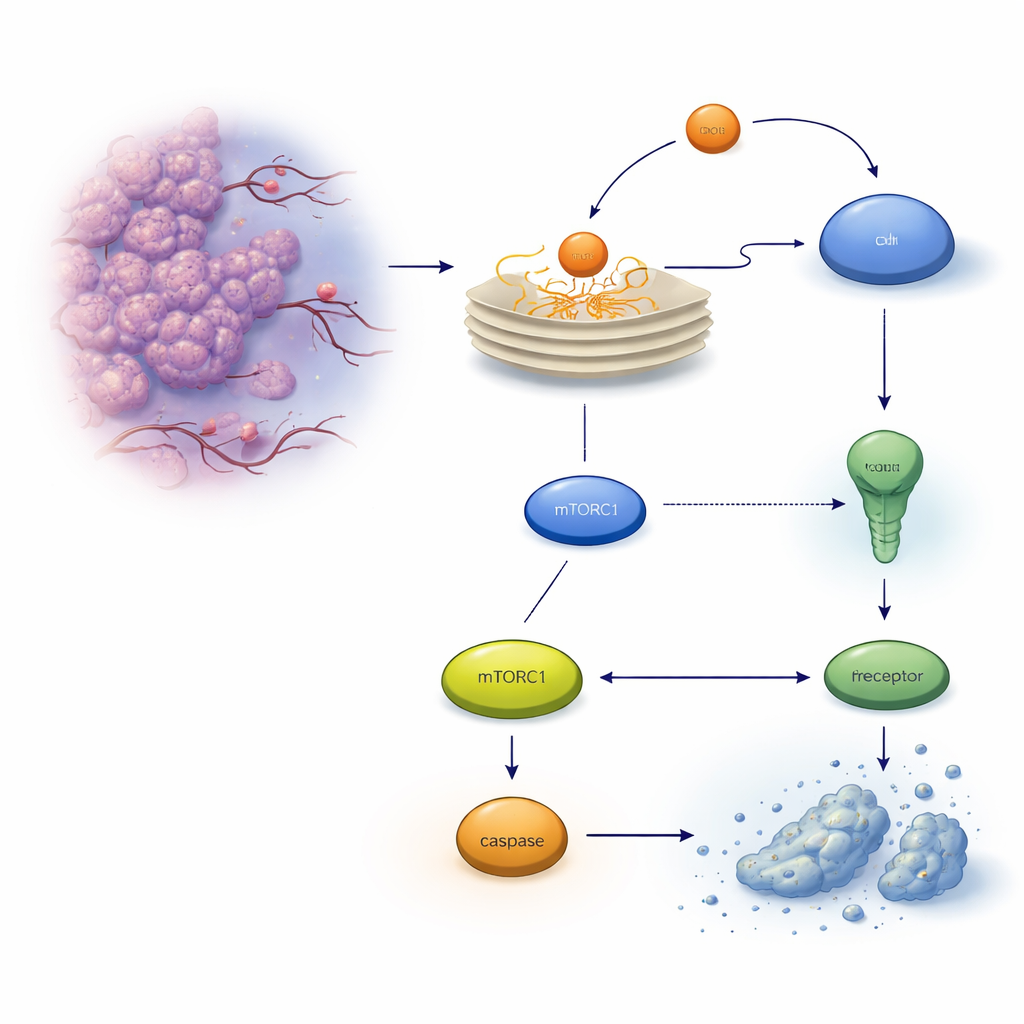
Ukryty zawór bezpieczeństwa wewnątrz komórek guza
Kiedy białka nie są prawidłowo fałdowane, gromadzą się w retikulum endoplazmatycznym (ER), uruchamiając system alarmowy zwany odpowiedzią na nieprawidłowo sfałdowane białka. Początkowo ta odpowiedź próbuje przywrócić równowagę, ale jeśli stres jest zbyt silny lub trwa zbyt długo, może przełączyć się w program samozniszczenia zwany apoptozą. W komórkach raka jelita grubego jednym z kluczowych wykonawców jest receptor śmierci na powierzchni komórki, TRAILR2/DR5, który może aktywować enzym tnący białka, kaspazę‑8, i ostatecznie doprowadzić do śmierci komórki. Autorzy skupili się na innym białku indukowanym przez stres, REDD1/DDIT4, już znanym z pomagania komórkom w adaptacji do trudnych warunków, i sprawdzili, czy wpływa ono na tę decyzję o życiu i śmierci.
Jak białko stresowe przechyla równowagę na stronę przeżycia
Naukowcy wystawili linie ludzkich komórek raka jelita grubego na leki wywołujące stres ER i śledzili poziomy REDD1 w czasie. Odkryli, że stres ER szybko zwiększa produkcję REDD1 w uporządkowany sposób kontrolowany przez inny czynnik reagujący na stres, ATF4. Obecność REDD1 tłumiła aktywność mTORC1, głównego regulatora wzrostu i syntezy białek. Wyciszenie lub usunięcie REDD1 za pomocą interferencji RNA lub edycji genów CRISPR znacznie zwiększało podatność komórek nowotworowych na apoptozę podczas stresu ER, a ten dodatkowy rozpad komórek można było zablokować inhibitorami kaspaz lub farmakologicznym wyłączeniem mTORC1, co pokazuje, że REDD1 zwykle chroni komórki poprzez powstrzymywanie tej ścieżki wzrostu w warunkach stresu.
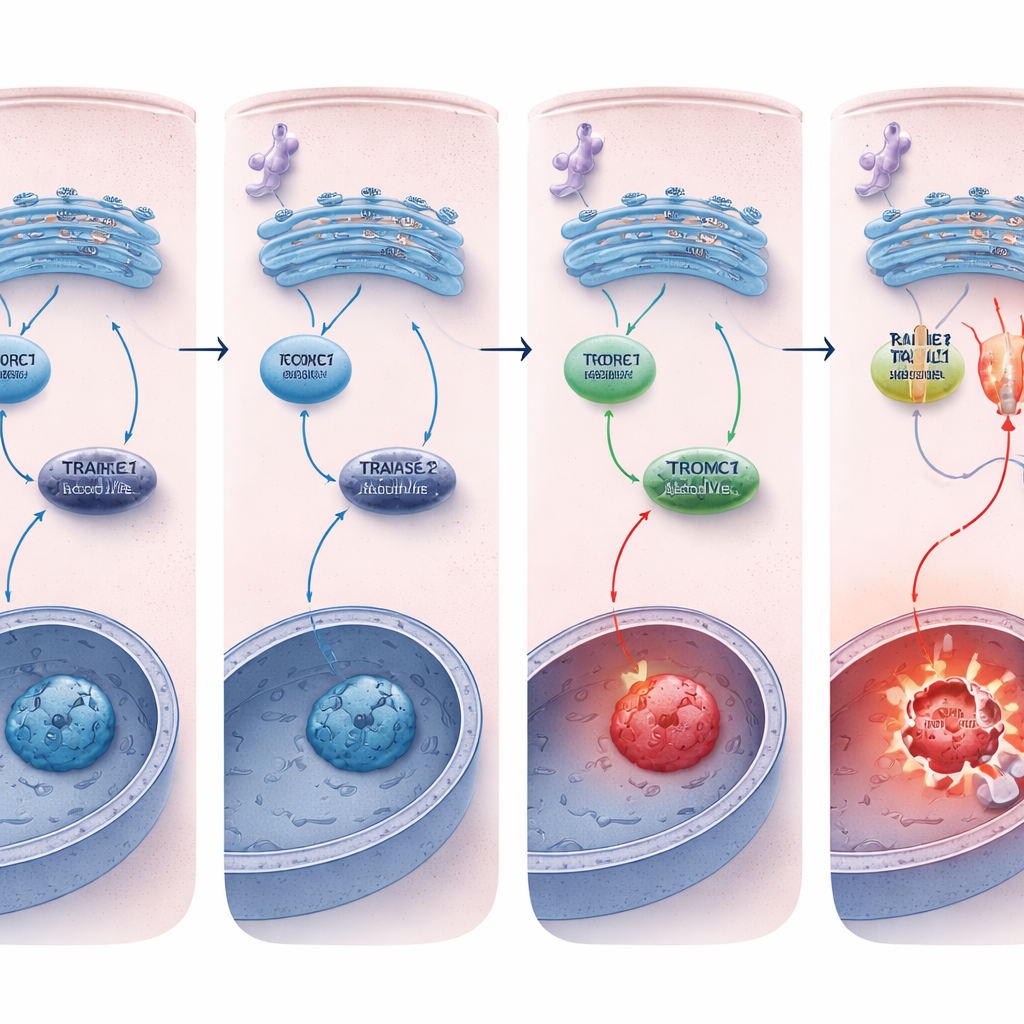
Utrzymywanie wyłączonego przełącznika śmierci
Dlaczego komórki pozbawione REDD1 ginęły łatwiej? Kluczową różnicą był receptor śmierci TRAILR2/DR5. Pod przewlekłym stresem ER zarówno komórki normalne, jak i pozbawione REDD1 aktywowały w podobnym stopniu wstępne szlaki stresowe, ale komórki bez REDD1 wykazywały znacznie większy wzrost TRAILR2/DR5 na poziomie RNA i białka, wraz z silniejszą aktywacją kaspazy‑8. Wyciszenie TRAILR2/DR5, ale nie pokrewnego receptora, znacząco zmniejszało apoptozę w tych komórkach pozbawionych REDD1, dowodząc, że nasilona odpowiedź śmierci była napędzana głównie przez ten receptor. W trójwymiarowych sferoidach nowotworowych, które lepiej naśladują rzeczywiste guzy i naturalnie gromadzą stres, poziomy REDD1 były już wysokie, a aktywność mTORC1 niska, a usunięcie REDD1 ponownie czyniło te mini‑guzy znacznie bardziej podatnymi na śmierć komórkową wywołaną stresem ER.
Rola hamulca transkrypcyjnego
Aby zrozumieć, jak REDD1 kontroluje TRAILR2/DR5, zespół zastosował sekwencjonowanie RNA, by wyszukać regulatorów, których poziomy zmieniały się po utracie REDD1. Wskazali czynnik transkrypcyjny zwany EVI‑1/MECOM, wcześniej powiązany z wzrostem nowotworu i represją genów. W komórkach pozbawionych REDD1 poziomy EVI‑1/MECOM spadły. Redukcja EVI‑1/MECOM w komórkach normalnych zwiększała ich wrażliwość na stres ER, podwyższała ekspresję TRAILR2/DR5 i wywoływała apoptozę zależną od kaspazy‑8, naśladując efekt utraty REDD1. Autorzy odkryli również, że korepresory zwane CtBP1/2, znane partnerki EVI‑1/MECOM w niektórych nowotworach, mogły dodatkowo regulować poziomy TRAILR2/DR5 i wrażliwość na stres w sposób zależny od typu komórek. Razem te wyniki sugerują, że REDD1 pomaga utrzymać represję genu TRAILR2/DR5 opartą na EVI‑1/MECOM, opóźniając aktywację przełącznika śmierci podczas przedłużonego stresu.
Co to oznacza dla przyszłego leczenia nowotworów
Dla czytelnika nieznającego tematu główny wniosek jest taki, że komórki raka jelita grubego posiadają wewnętrzny mechanizm pozwalający im „przetrwać” ciężki stres białkowy zamiast umierać, gdy powinny. Białko REDD1 działa jak zawór bezpieczeństwa: wycisza główną ścieżkę wzrostu i podtrzymuje system represji genów, który powstrzymuje mocny receptor śmierci TRAILR2/DR5 przed włączeniem się zbyt wcześnie. Gdy REDD1 lub jego partner EVI‑1/MECOM są usunięci, zestresowane komórki nowotworowe znacznie częściej ulegają samozniszczeniu. Praca ta sugeruje, że celowanie w REDD1 lub jego represory pośrednie mogłoby pozbawić guzy tej ochronnej poduszki, czyniąc komórki nowotworowe w nieprzyjaznych, słabo odżywionych obszarach guza bardziej podatnymi na terapie, które zwiększają stres ER i popychają komórki w stronę apoptozy.
Cytowanie: Mora-Molina, R., El Yousfi, Y., Hagenlocher, C. et al. REDD1/DDIT4 counteracts endoplasmic reticulum stress-induced apoptosis by controlling the expression of death receptor TRAILR2/DR5 in cancer cells. Cell Death Dis 17, 425 (2026). https://doi.org/10.1038/s41419-026-08648-7
Słowa kluczowe: stres ER, rak jelita grubego, apoptoza, TRAILR2/DR5, REDD1/DDIT4