Clear Sky Science · de
REDD1/DDIT4 wirkt dem endoplasmatischen Retikulum-Stress-induzierten Zelltod entgegen, indem es die Expression des Todesrezeptors TRAILR2/DR5 in Krebszellen kontrolliert
Warum gestresste Tumorzellen wichtig sind
Krebszellen wachsen nicht in Komfort. Innerhalb eines Tumors sind Zellen niedriger Sauerstoffversorgung, knappen Nährstoffen und einem Aufbau beschädigter Proteine ausgesetzt. Unter diesem Druck sterben einige Zellen, aber andere passen sich an und werden schwerer mit Therapien zu erreichen. Diese Arbeit untersucht, wie Kolorektalkarzinomzellen eine spezifische Form innerer Belastung — Probleme im endoplasmatischen Retikulum, der Proteinfaltungsfabrik der Zelle — überleben, und identifiziert eine molekulare „Bremse“, die verhindert, dass sich diese gestressten Zellen selbst zerstören.
Ein verstecktes Sicherheitsventil in Tumorzellen
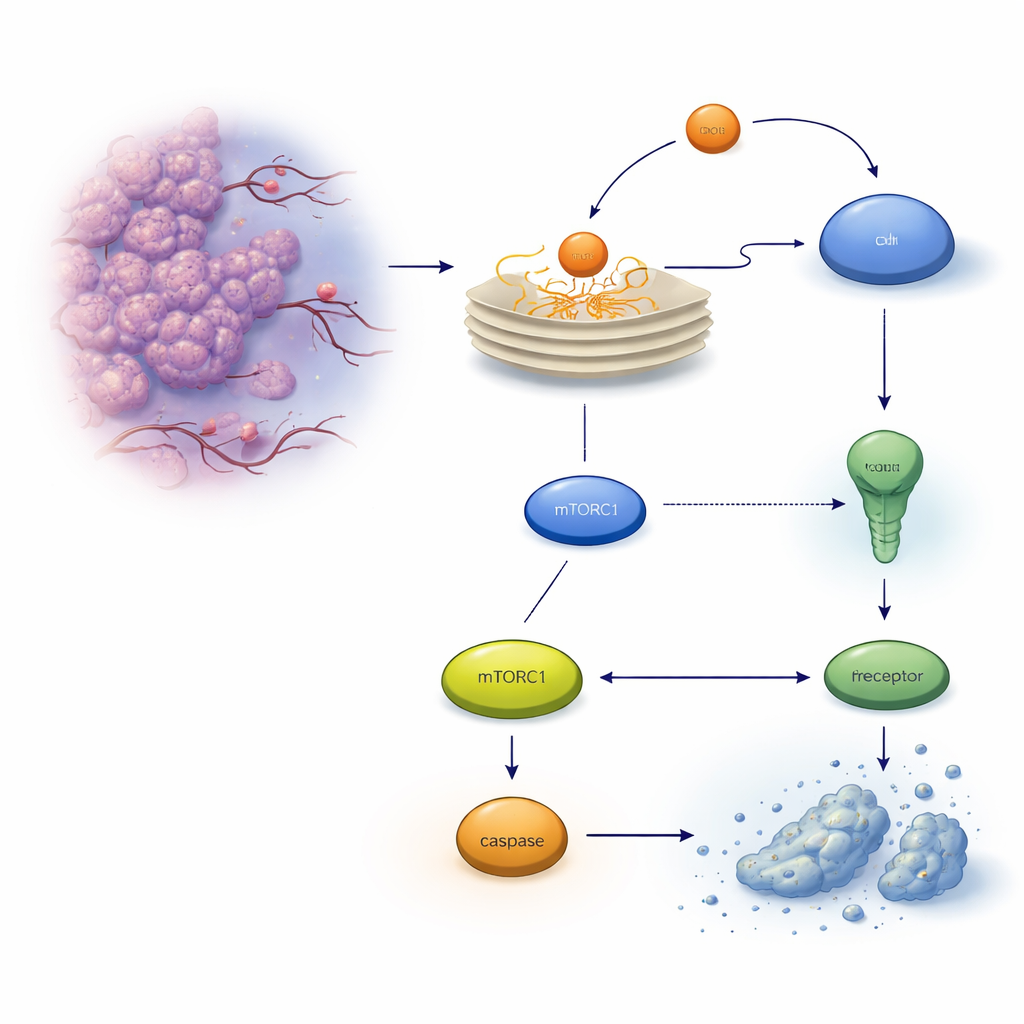
Wenn Proteine nicht korrekt gefaltet sind, sammeln sie sich im endoplasmatischen Retikulum (ER) an und lösen ein Alarmsystem aus, das als unfolded protein response bezeichnet wird. Zunächst versucht diese Antwort, das Gleichgewicht wiederherzustellen, doch wenn der Stress zu stark ist oder zu lange anhält, kann sie in ein Selbstzerstörungsprogramm umschalten, das als Apoptose bekannt ist. In Kolorektalkarzinomzellen ist ein wichtiger Ausführender ein Todesrezeptor an der Zelloberfläche namens TRAILR2/DR5, der ein proteolytisches Enzym, Caspase‑8, aktivieren und schließlich die Zelle abtöten kann. Die Autoren konzentrierten sich auf ein weiteres stressinduziertes Protein, REDD1/DDIT4, das bereits dafür bekannt ist, Zellen bei widrigen Bedingungen zu helfen, und fragten, ob es diese Lebens‑oder‑Todes‑Entscheidung beeinflusst.
Wie ein Stressprotein das Gleichgewicht zugunsten des Überlebens verschiebt
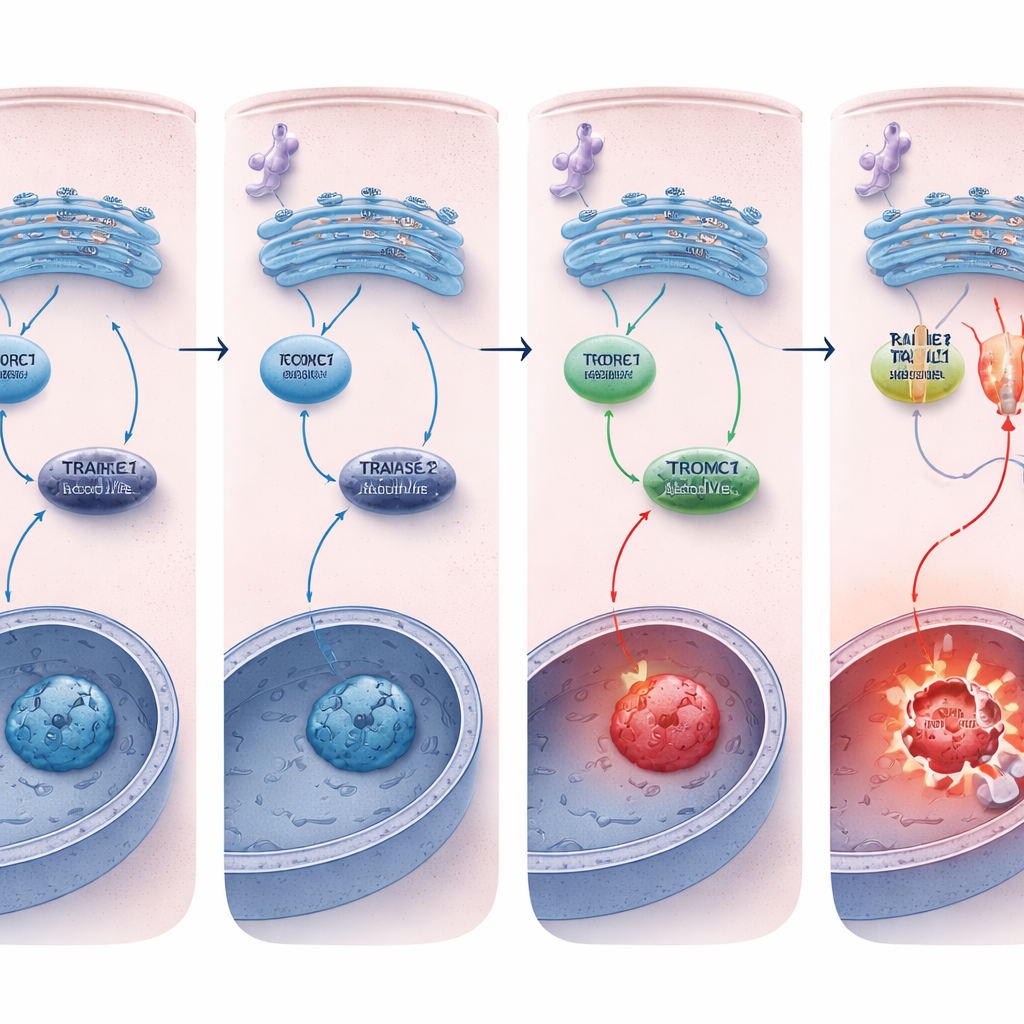
Die Forscher setzten menschliche Kolonkarzinomzelllinien Wirkstoffen aus, die ER‑Stress verursachen, und verfolgten die REDD1‑Spiegel über die Zeit. Sie fanden heraus, dass ER‑Stress die Produktion von REDD1 schnell erhöhte, in einer geordneten Abfolge, die von einem anderen stressreaktiven Faktor, ATF4, gesteuert wird. Wenn REDD1 vorhanden war, dämpfte es die Aktivität von mTORC1, einem wichtigen Regulator von Wachstum und Proteinsynthese. Das Herunterregulieren oder Entfernen von REDD1 mittels RNA‑Interferenz oder CRISPR‑Geneditierung machte die Krebszellen deutlich anfälliger für Apoptose unter ER‑Stress, und dieses zusätzliche Zellsterben ließ sich durch Caspase‑Hemmer oder durch pharmakologische Hemmung von mTORC1 blockieren, was zeigt, dass REDD1 Zellen normalerweise dadurch schützt, dass es diesen Wachstumsweg während Stress einschränkt.
Den Todes-Schalter heruntergeregelt halten
Warum starben REDD1‑defiziente Zellen leichter? Der entscheidende Unterschied lag beim Todesrezeptor TRAILR2/DR5. Unter chronischem ER‑Stress aktivierten sowohl normale als auch REDD1‑defiziente Zellen die upstream Stresswege ähnlich, aber Zellen ohne REDD1 zeigten einen deutlich stärkeren Anstieg von TRAILR2/DR5 auf RNA‑ und Proteinebene sowie eine stärkere Aktivierung von Caspase‑8. Das Stilllegen von TRAILR2/DR5, nicht jedoch eines verwandten Rezeptors, reduzierte die Apoptose in diesen REDD1‑defizienten Zellen stark und bewies, dass die verstärkte Todesantwort hauptsächlich über diesen Rezeptor vermittelt wurde. In dreidimensionalen Tumor‑Sphäroiden, die reale Tumore besser nachahmen und von Natur aus Stress akkumulieren, waren REDD1‑Spiegel bereits hoch und die mTORC1‑Aktivität niedrig; das Entfernen von REDD1 machte diese Mini‑Tumoren erneut wesentlich anfälliger für ER‑Stress‑induzierten Zelltod.
Die Rolle einer Transkriptionsbremse
Um zu verstehen, wie REDD1 TRAILR2/DR5 kontrolliert, nutzte das Team RNA‑Sequenzierung, um nach Regulatoren zu suchen, deren Spiegel sich beim Verlust von REDD1 änderten. Sie identifizierten einen Transkriptionsfaktor namens EVI‑1/MECOM, der zuvor mit Tumorwachstum und Genrepression in Verbindung gebracht worden war. In REDD1‑defizienten Zellen sanken die EVI‑1/MECOM‑Spiegel. Das Reduzieren von EVI‑1/MECOM in ansonsten normalen Zellen machte diese empfindlicher gegenüber ER‑Stress, erhöhte die TRAILR2/DR5‑Expression und führte zu Caspase‑8‑abhängiger Apoptose, was den Effekt des REDD1‑Verlusts nachahmte. Die Autoren fanden außerdem, dass Co‑Repressoren namens CtBP1/2, bekannte Partner von EVI‑1/MECOM in einigen Tumoren, TRAILR2/DR5‑Spiegel und Stressempfindlichkeit weiter in einer zelltypspezifischen Weise feinabstimmen konnten. Zusammengenommen deuten diese Ergebnisse darauf hin, dass REDD1 hilft, die EVI‑1/MECOM‑basierte Repression des TRAILR2/DR5‑Gens aufrechtzuerhalten und so die Aktivierung des Todes‑Schalters während anhaltendem Stress verzögert.
Was das für die künftige Krebstherapie bedeutet
Für ein Laienpublikum ist die wichtigste Erkenntnis, dass Kolonkarzinomzellen einen internen Mechanismus besitzen, der es ihnen erlaubt, schweren Proteinstress „auszusitzen“, anstatt zu sterben, wenn sie es sollten. Das Protein REDD1 wirkt wie ein Sicherheitsventil: Es dämpft einen wichtigen Wachstumsweg und stützt ein Genrepressionssystem, das einen potenten Todesrezeptor, TRAILR2/DR5, davon abhält, zu früh anzuspringen. Wenn REDD1 oder sein Partner EVI‑1/MECOM entfernt werden, neigen gestresste Krebszellen deutlich eher zur Selbstzerstörung. Diese Arbeit legt nahe, dass das gezielte Angreifen von REDD1 oder seiner nachgeschalteten Repressoren Tumoren diesen Schutzpuffer entziehen könnte, sodass Krebszellen in feindlichen, schlecht genährten Bereichen eines Tumors anfälliger für Therapien werden, die ER‑Stress erhöhen und Zellen in Richtung Apoptose treiben.
Zitation: Mora-Molina, R., El Yousfi, Y., Hagenlocher, C. et al. REDD1/DDIT4 counteracts endoplasmic reticulum stress-induced apoptosis by controlling the expression of death receptor TRAILR2/DR5 in cancer cells. Cell Death Dis 17, 425 (2026). https://doi.org/10.1038/s41419-026-08648-7
Schlüsselwörter: ER-Stress, kolorektales Karzinom, Apoptose, TRAILR2/DR5, REDD1/DDIT4