Clear Sky Science · it
REDD1/DDIT4 contrasta l’apoptosi indotta dallo stress del reticolo endoplasmatico controllando l’espressione del recettore della morte TRAILR2/DR5 nelle cellule tumorali
Perché le cellule tumorali sottoposte a stress sono importanti
Le cellule cancerose non crescono in condizioni confortevoli. All’interno di un tumore le cellule affrontano basso ossigeno, nutrienti scarsi e l’accumulo di proteine danneggiate. Sotto questa pressione alcune cellule muoiono, ma altre si adattano e diventano più difficili da eliminare con le terapie. Questo studio esplora come le cellule del cancro del colon sopravvivano a un tipo specifico di stress interno — il malfunzionamento del reticolo endoplasmatico, la “fabbrica” di ripiegamento delle proteine della cellula — e identifica un “freno” molecolare che impedisce a queste cellule stressate di autodemolirsi.
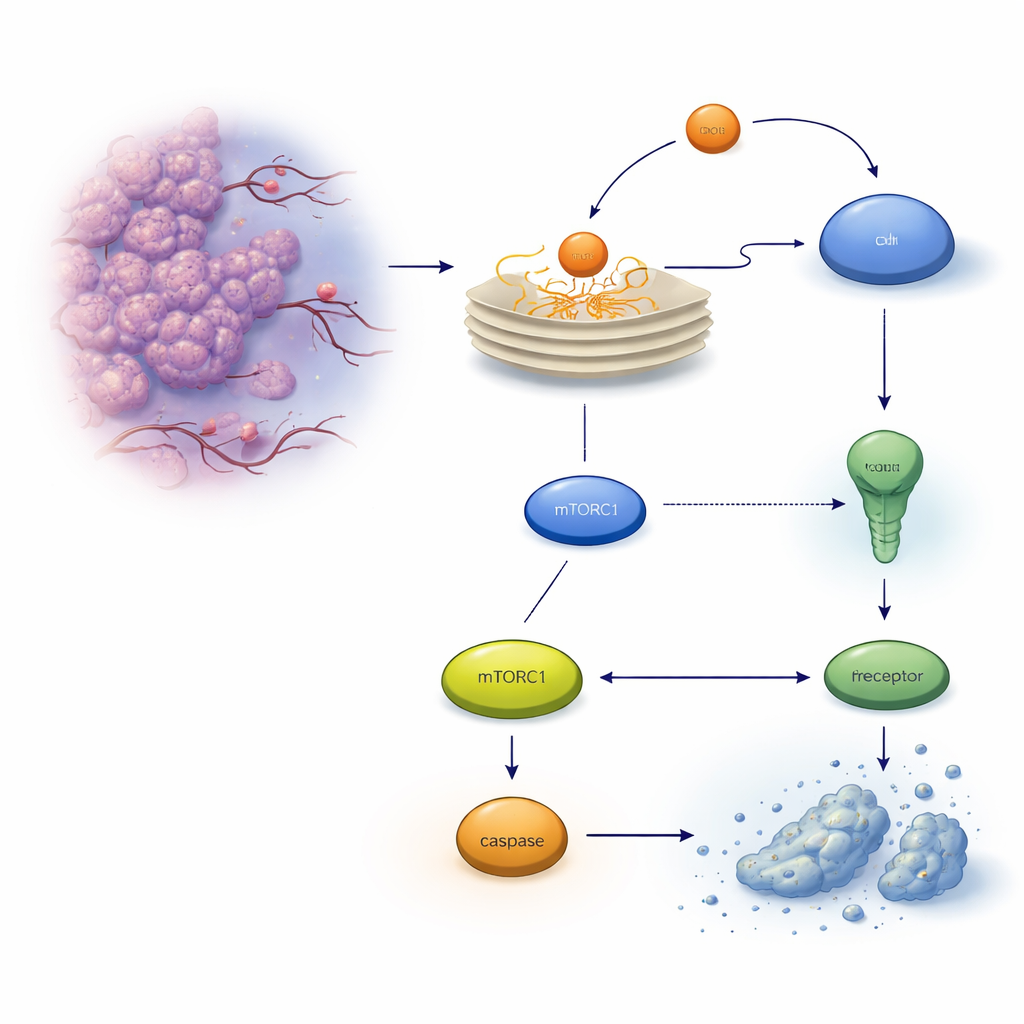
Una valvola di sicurezza nascosta nelle cellule tumorali
Quando le proteine non si ripiegano correttamente, si accumulano nel reticolo endoplasmatico (RE), attivando un sistema di allarme chiamato risposta alle proteine non ripiegate. All’inizio questa risposta cerca di ristabilire l’equilibrio, ma se lo stress è troppo intenso o prolungato può ribaltarsi in un programma di autodistruzione noto come apoptosi. Nelle cellule del cancro del colon, un esecutore chiave è un recettore della morte sulla superficie cellulare chiamato TRAILR2/DR5, che può attivare un enzima che taglia le proteine, caspasi‑8, e in ultima analisi uccidere la cellula. Gli autori si sono concentrati su un’altra proteina indotta dallo stress, REDD1/DDIT4, nota per aiutare le cellule ad adattarsi a condizioni avverse, e hanno chiesto se influisca su questa decisione di vita o di morte.
Come una proteina da stress inclina la bilancia verso la sopravvivenza
I ricercatori hanno esposto linee cellulari umane di cancro del colon a farmaci che inducono stress del RE e hanno monitorato i livelli di REDD1 nel tempo. Hanno osservato che lo stress del RE aumentava rapidamente la produzione di REDD1 in modo ordinato, controllata da un altro fattore sensibile allo stress, ATF4. Quando REDD1 era presente, attenuava l’attività di mTORC1, un importante regolatore della crescita e della sintesi proteica. L’abbattimento o la delezione di REDD1 mediante interferenza con RNA o modifica genica CRISPR rendeva le cellule tumorali molto più propense ad andare incontro ad apoptosi sotto stress del RE, e questa morte cellulare aggiuntiva poteva essere bloccata da inibitori delle caspasi o dallo spegnimento farmacologico di mTORC1, dimostrando che REDD1 protegge normalmente le cellule limitando questa via di crescita durante lo stress.
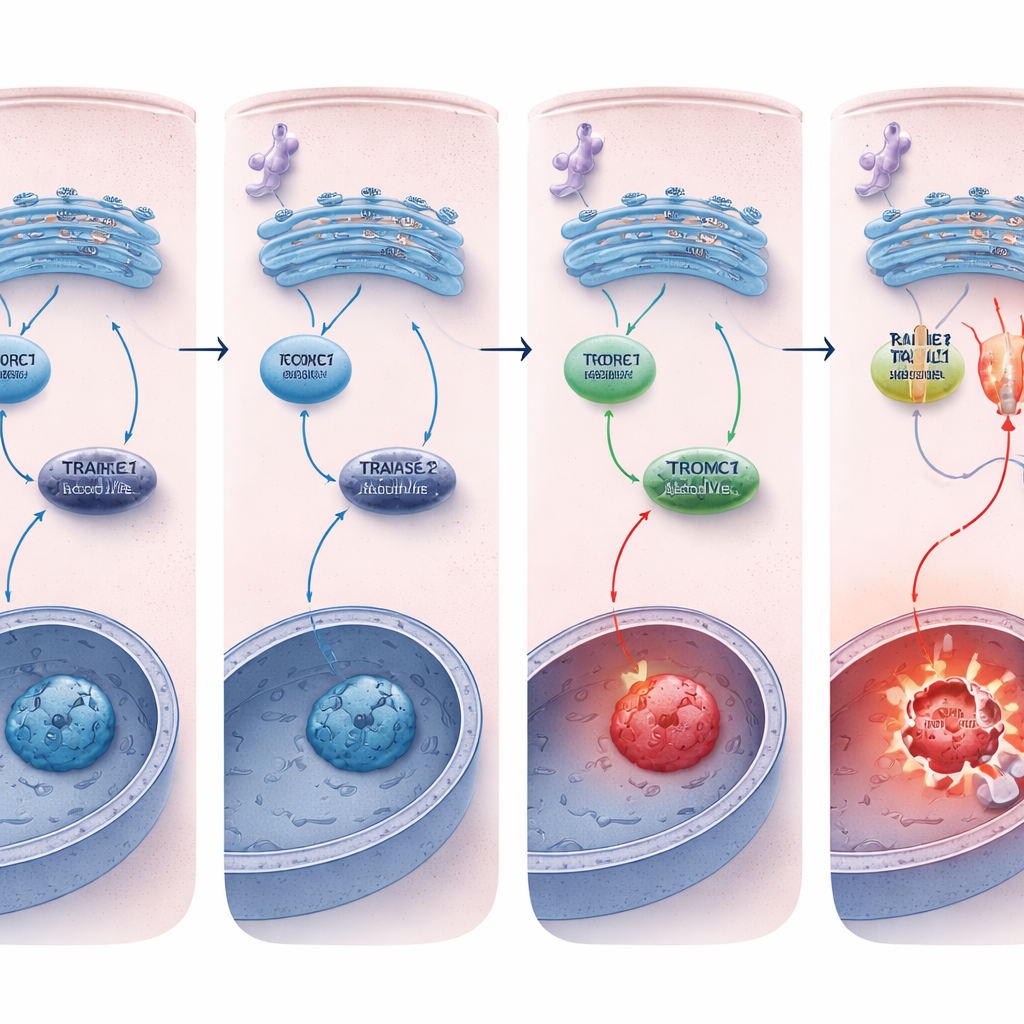
Mantenere il selettore della morte abbassato
Perché le cellule prive di REDD1 morivano più facilmente? La differenza chiave riguardava il recettore della morte TRAILR2/DR5. In condizioni di stress cronico del RE, sia le cellule normali sia quelle prive di REDD1 attivavano in modo simile le vie di stress a monte, ma le cellule senza REDD1 mostravano un aumento molto maggiore di TRAILR2/DR5 sia a livello di RNA sia di proteina, insieme a una più forte attivazione di caspasi‑8. Silenziare TRAILR2/DR5, ma non un recettore correlato, riduceva nettamente l’apoptosi in queste cellule prive di REDD1, dimostrando che la risposta di morte accentuata era guidata principalmente attraverso questo recettore. In sfere tumorali tridimensionali, che imitano meglio i tumori reali e accumulano naturalmente stress, i livelli di REDD1 erano già elevati e l’attività di mTORC1 bassa, e la rimozione di REDD1 rendeva di nuovo questi mini‑tumori molto più vulnerabili alla morte cellulare indotta dallo stress del RE.
Il ruolo di un freno trascrizionale
Per comprendere come REDD1 controlli TRAILR2/DR5, il gruppo ha usato il sequenziamento dell’RNA per cercare regolatori i cui livelli cambiavano quando REDD1 veniva perso. Hanno identificato un fattore di trascrizione chiamato EVI‑1/MECOM, precedentemente collegato alla crescita tumorale e alla repressione genica. Nelle cellule prive di REDD1, i livelli di EVI‑1/MECOM diminuivano. Ridurre EVI‑1/MECOM in cellule altrimenti normali le rendeva più sensibili allo stress del RE, aumentava l’espressione di TRAILR2/DR5 e induceva apoptosi dipendente da caspasi‑8, imitando l’effetto della perdita di REDD1. Gli autori hanno anche rilevato che co‑repressori chiamati CtBP1/2, partner noti di EVI‑1/MECOM in alcuni tumori, potevano modulare ulteriormente i livelli di TRAILR2/DR5 e la sensibilità allo stress in modo specifico per tipo cellulare. Nel complesso, questi risultati suggeriscono che REDD1 aiuta a mantenere la repressione del gene TRAILR2/DR5 mediata da EVI‑1/MECOM, ritardando l’attivazione del selettore della morte durante lo stress prolungato.
Cosa significa per il futuro trattamento del cancro
Per un pubblico non specialistico, la principale conclusione è che le cellule del cancro del colon possiedono un meccanismo interno che permette loro di “sopportare” uno stress proteico severo invece di morire quando dovrebbero. La proteina REDD1 funziona come una valvola di sicurezza: attenua una via di crescita importante e supporta un sistema di repressione genica che impedisce l’attivazione prematura di un potente recettore della morte, TRAILR2/DR5. Quando REDD1 o il suo partner EVI‑1/MECOM vengono rimossi, le cellule tumorali stressate sono molto più propense ad autodestruirsi. Questo lavoro suggerisce che mirare a REDD1 o ai suoi repressori downstream potrebbe privare i tumori di questo cuscinetto protettivo, rendendo le cellule cancerose in regioni ostili e povere di nutrienti di un tumore più vulnerabili alle terapie che aumentano lo stress del RE e spingono le cellule verso l’apoptosi.
Citazione: Mora-Molina, R., El Yousfi, Y., Hagenlocher, C. et al. REDD1/DDIT4 counteracts endoplasmic reticulum stress-induced apoptosis by controlling the expression of death receptor TRAILR2/DR5 in cancer cells. Cell Death Dis 17, 425 (2026). https://doi.org/10.1038/s41419-026-08648-7
Parole chiave: Stress del RE, cancro del colon-retto, apoptosi, TRAILR2/DR5, REDD1/DDIT4