Clear Sky Science · en
REDD1/DDIT4 counteracts endoplasmic reticulum stress-induced apoptosis by controlling the expression of death receptor TRAILR2/DR5 in cancer cells
Why stressed tumor cells matter
Cancer cells don’t grow in comfort. Inside a tumor, cells face low oxygen, scarce nutrients and a buildup of damaged proteins. Under this pressure, some cells die, but others adapt and become harder to kill with treatment. This paper explores how colon cancer cells survive one specific kind of internal stress — trouble in the endoplasmic reticulum, the cell’s protein-folding factory — and identifies a molecular “brake” that keeps these stressed cells from self-destructing.
A hidden safety valve inside tumor cells
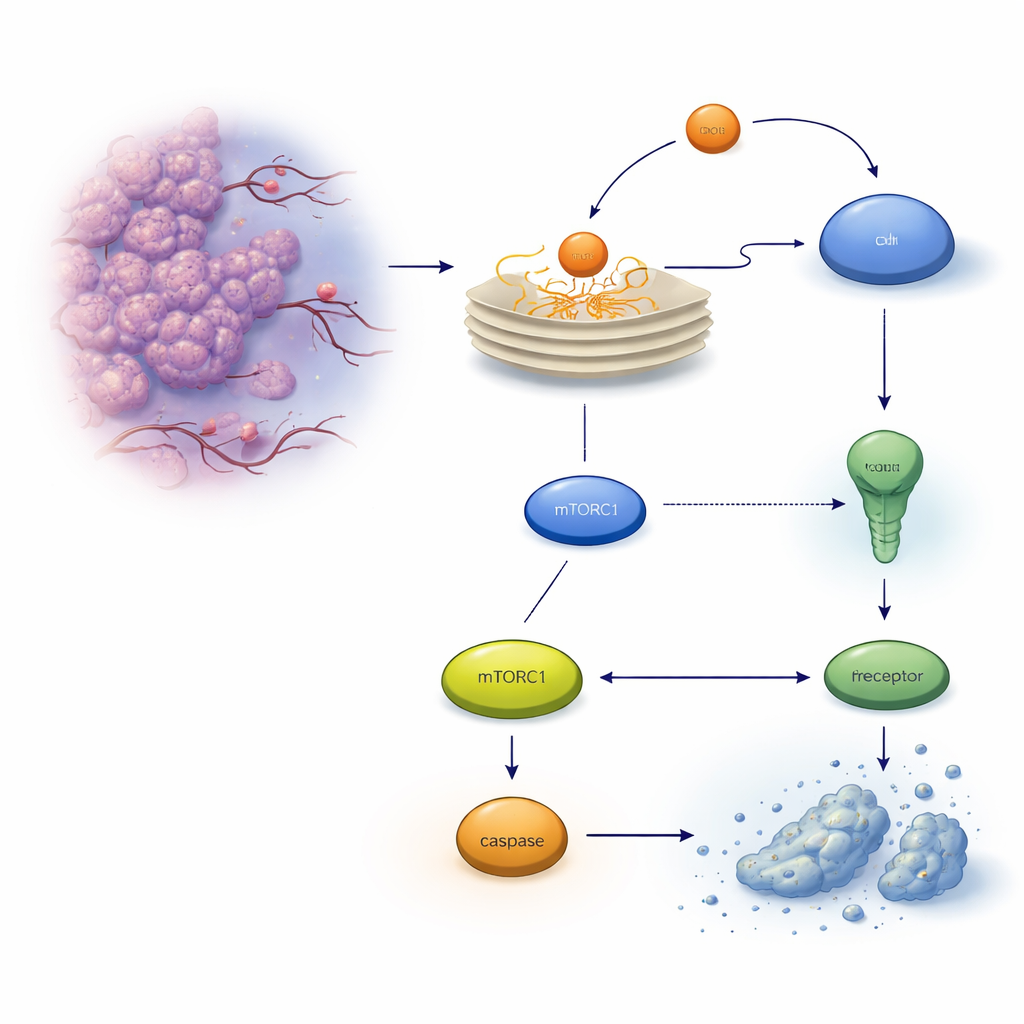
When proteins are not folded properly, they accumulate in the endoplasmic reticulum (ER), triggering an alarm system called the unfolded protein response. At first, this response tries to restore balance, but if stress is too strong or lasts too long, it can flip into a self-destruct program known as apoptosis. In colon cancer cells, one key executioner is a death receptor on the cell surface called TRAILR2/DR5, which can activate a protein-cutting enzyme, caspase-8, and ultimately kill the cell. The authors focused on another stress‑induced protein, REDD1/DDIT4, already known to help cells adapt to harsh conditions, and asked whether it influences this life‑or‑death decision.
How a stress protein tilts the balance toward survival
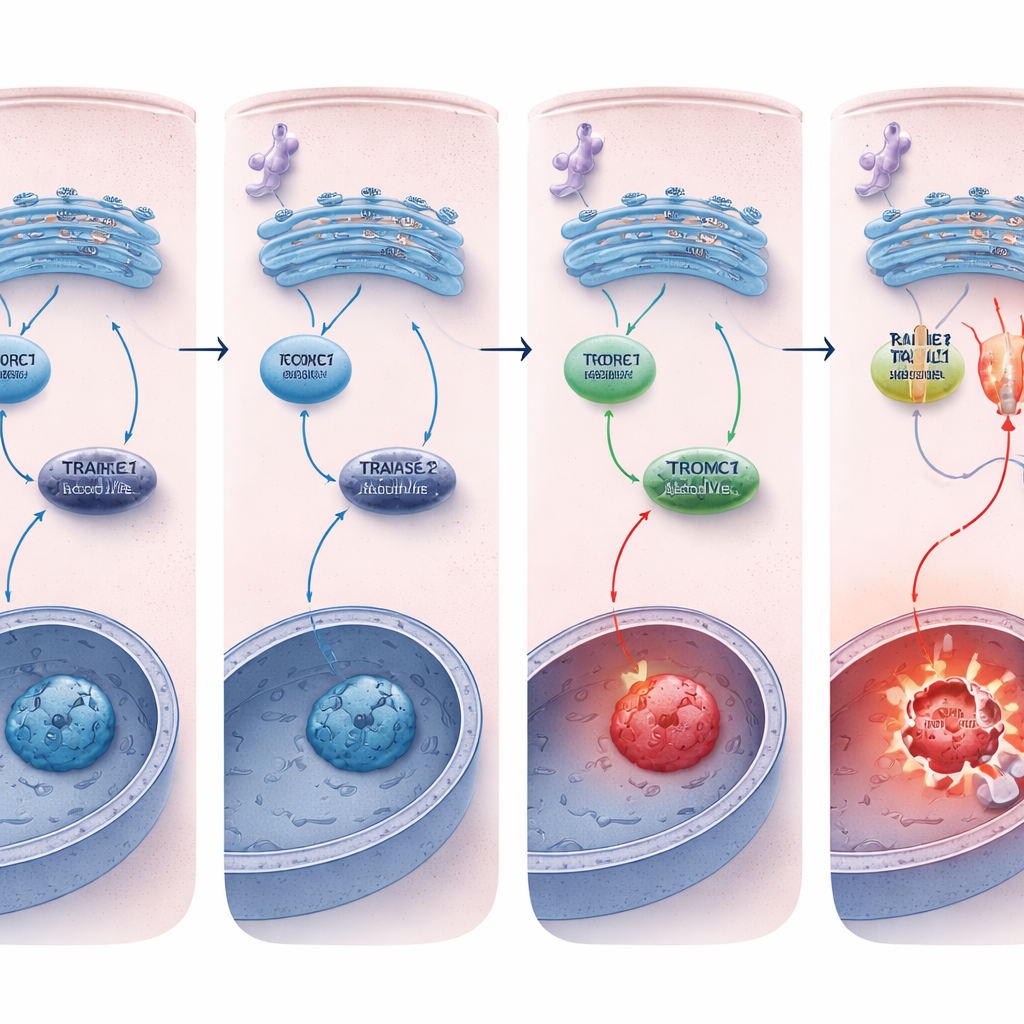
The researchers exposed human colon cancer cell lines to drugs that cause ER stress and tracked REDD1 levels over time. They found that ER stress rapidly boosted REDD1 production in an orderly fashion controlled by another stress‑responsive factor, ATF4. When REDD1 was present, it dampened the activity of mTORC1, a major growth and protein‑synthesis regulator. Knocking down or deleting REDD1 using RNA interference or CRISPR gene editing made the cancer cells much more likely to undergo apoptosis under ER stress, and this extra cell death could be blocked by caspase inhibitors or by pharmacologically shutting down mTORC1, showing that REDD1 normally protects cells by restraining this growth pathway during stress.
Keeping the death switch turned down
Why did REDD1‑deficient cells die more easily? The key difference was in the death receptor TRAILR2/DR5. Under chronic ER stress, both normal and REDD1‑deficient cells activated upstream stress pathways similarly, but cells lacking REDD1 showed a much larger rise in TRAILR2/DR5 at both RNA and protein levels, along with stronger activation of caspase‑8. Silencing TRAILR2/DR5, but not a related receptor, sharply reduced apoptosis in these REDD1‑deficient cells, proving that the heightened death response was driven mainly through this receptor. In three‑dimensional tumor spheroids, which better mimic real tumors and naturally accumulate stress, REDD1 levels were already high and mTORC1 activity was low, and removing REDD1 again made these mini‑tumors far more vulnerable to ER‑stress‑induced cell death.
The role of a transcriptional brake
To understand how REDD1 controls TRAILR2/DR5, the team used RNA sequencing to search for regulators whose levels changed when REDD1 was lost. They pinpointed a transcription factor called EVI‑1/MECOM, previously linked to cancer growth and gene repression. In REDD1‑deficient cells, EVI‑1/MECOM levels dropped. Reducing EVI‑1/MECOM in otherwise normal cells made them more sensitive to ER stress, increased TRAILR2/DR5 expression and drove caspase‑8‑dependent apoptosis, mimicking the effect of losing REDD1. The authors also found that co‑repressors called CtBP1/2, known partners of EVI‑1/MECOM in some tumors, could further tune TRAILR2/DR5 levels and stress sensitivity in a cell‑type‑specific way. Together, these results suggest that REDD1 helps maintain EVI‑1/MECOM‑based repression of the TRAILR2/DR5 gene, delaying activation of the death switch during prolonged stress.
What this means for future cancer treatment
For a lay audience, the main takeaway is that colon cancer cells possess an internal mechanism that lets them “ride out” severe protein stress instead of dying when they should. The protein REDD1 acts like a safety valve: it quiets a major growth pathway and supports a gene‑repression system that keeps a potent death receptor, TRAILR2/DR5, from turning on too soon. When REDD1 or its partner EVI‑1/MECOM are removed, stressed cancer cells are far more likely to self‑destruct. This work suggests that targeting REDD1 or its downstream repressors might strip tumors of this protective buffer, making cancer cells in hostile, poorly nourished regions of a tumor more vulnerable to therapies that increase ER stress and push cells toward apoptosis.
Citation: Mora-Molina, R., El Yousfi, Y., Hagenlocher, C. et al. REDD1/DDIT4 counteracts endoplasmic reticulum stress-induced apoptosis by controlling the expression of death receptor TRAILR2/DR5 in cancer cells. Cell Death Dis 17, 425 (2026). https://doi.org/10.1038/s41419-026-08648-7
Keywords: ER stress, colorectal cancer, apoptosis, TRAILR2/DR5, REDD1/DDIT4