Clear Sky Science · pt
Estrutura multimodal para a análise conjunta de RNA de célula única e dados de sequenciamento do receptor de células T prevê a resposta de células T à imunoterapia do câncer
Por que prever a resposta imune é importante
Imunoterapias contra o câncer, como os inibidores de checkpoint, funcionam ao liberar as próprias células T do paciente para atacar tumores. Ainda assim, apenas alguns pacientes se beneficiam, e os médicos dispõem hoje de ferramentas limitadas para prever quem responderá. Este estudo apresenta uma estrutura computacional chamada TRIM que aprende a partir de medições detalhadas de células T em nível de célula única, tanto no sangue quanto nos tumores. Ao combinar informações sobre a atividade gênica de cada célula e seu receptor exclusivo, o TRIM pode prever como as células T vão se expandir e mudar após o tratamento, oferecendo uma forma potencial de antecipar o sucesso do tratamento a partir de uma coleta de sangue de rotina.
Olhando dentro de células T individuais
As células T patrulham o corpo em busca de sinais de perigo por meio de seus receptores, que reconhecem fragmentos moleculares específicos. Quando uma célula T encontra câncer, ela pode se multiplicar em um clone de muitas células quase idênticas e assumir papéis diferentes, desde assassinas de tumor ativas até células exauridas que não conseguem mais combater efetivamente. Tecnologias modernas de célula única agora podem ler, para cada célula T, tanto seu perfil de expressão gênica (quais genes estão ligados ou desligados) quanto a sequência de seu receptor. Os autores aplicaram essas ferramentas em milhares de células T de pacientes com câncer de cabeça e pescoço, colorretal e outros tipos, amostrando tanto sangue quanto tecido tumoral antes e depois da imunoterapia.
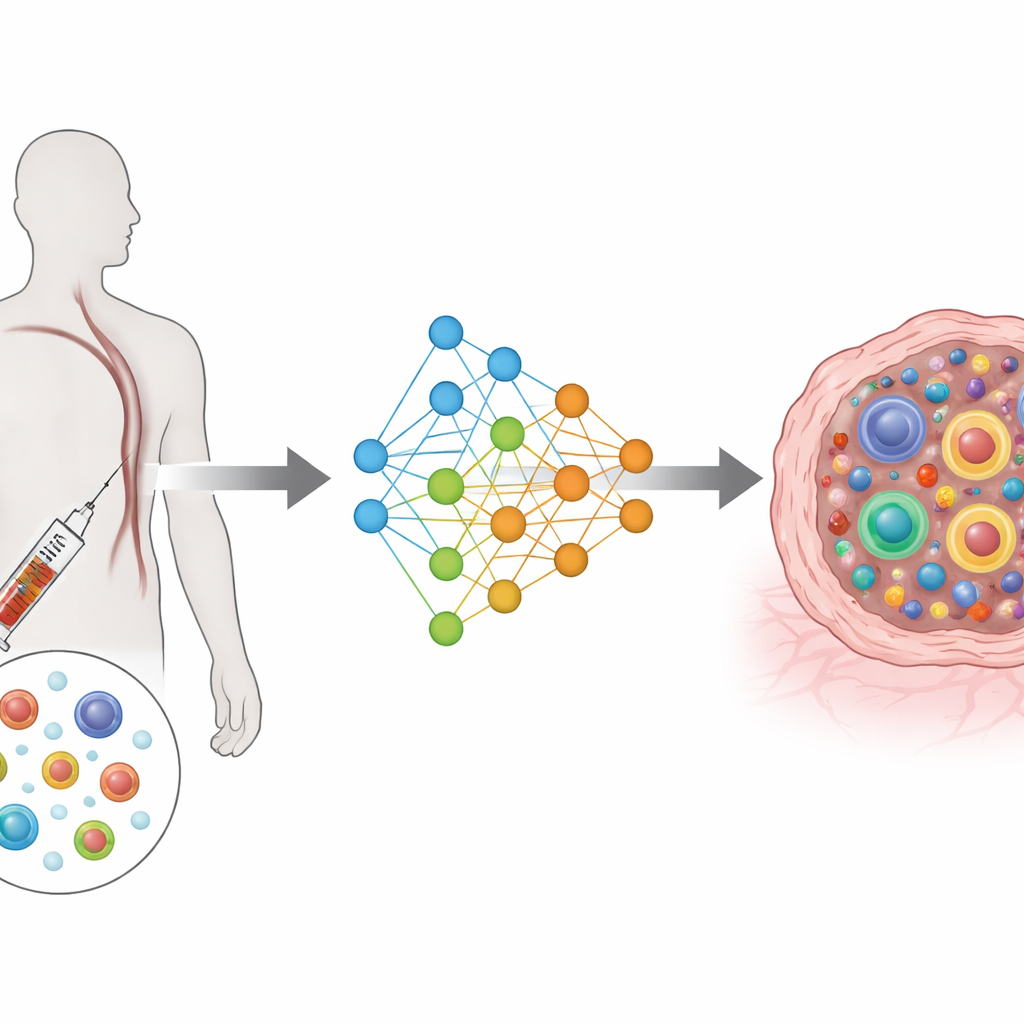
De medições complexas para um mapa compartilhado
O TRIM é construído sobre um tipo de arquitetura de aprendizado profundo conhecida como autoencoder variacional condicional. Em termos simples, ele comprime a informação rica de genes e receptores de cada célula em um mapa compartilhado de baixa dimensão, levando em conta quem é o paciente, de onde a célula veio (sangue ou tumor) e quando foi coletada (antes ou depois do tratamento). Módulos de entrada separados leem a atividade gênica e as características do receptor, e módulos de saída distintos tentam reconstruí-las. Uma função de perda especial aproxima células que pertencem ao mesmo clone nesse mapa, mantendo clones diferentes separados. Esse desenho foca o modelo no que é mais relevante biologicamente: o tamanho de cada clone e como suas células se distribuem entre estados funcionais.
O que liga receptores, atividade gênica e clones
Ao comparar sistematicamente a similaridade de receptores e a expressão gênica através de múltiplos conjuntos de dados, os pesquisadores descobriram que células que compartilham exatamente o mesmo receptor são, de fato, mais semelhantes entre si na expressão gênica do que células T aleatórias, especialmente para clones grandes e expandidos. Entretanto, além de correspondências exatas, pequenas alterações na sequência do receptor não acompanham de forma confiável o quanto duas células são semelhantes na expressão gênica. Em outras palavras, células com receptores quase idênticos podem parecer tão diferentes quanto células com receptores completamente não relacionados. Isso levou a equipe a concentrar-se na identidade do clone e no tamanho do clone, em vez da similaridade fina da sequência do receptor, como a ponte crucial entre a informação do receptor e o estado da célula.
Prevendo células T ligadas ao tumor a partir do sangue
O teste central para o TRIM foi se ele conseguiria usar apenas dados de sangue pré-tratamento de um paciente para gerar previsões realistas das células T desse paciente no tumor e no sangue após a terapia. Os autores treinaram o TRIM em todos os pacientes menos um e então pediram que previsse o paciente deixado de fora, repetindo isso entre as coortes. Eles mostraram que o modelo captura com precisão o panorama geral dos estados gênicos das células T e a diversidade de clones entre condições. Mais marcante, para clones de receptores individuais presentes antes da terapia, o TRIM pôde prever quais se expandiriam após o tratamento com alta acurácia, superando claramente abordagens padrão de aprendizado de máquina. Também reproduziu assinaturas conhecidas de resposta bem-sucedida, como o aumento de células CD8 ativadas e em proliferação e programas gênicos específicos associados à efetiva eliminação tumoral.
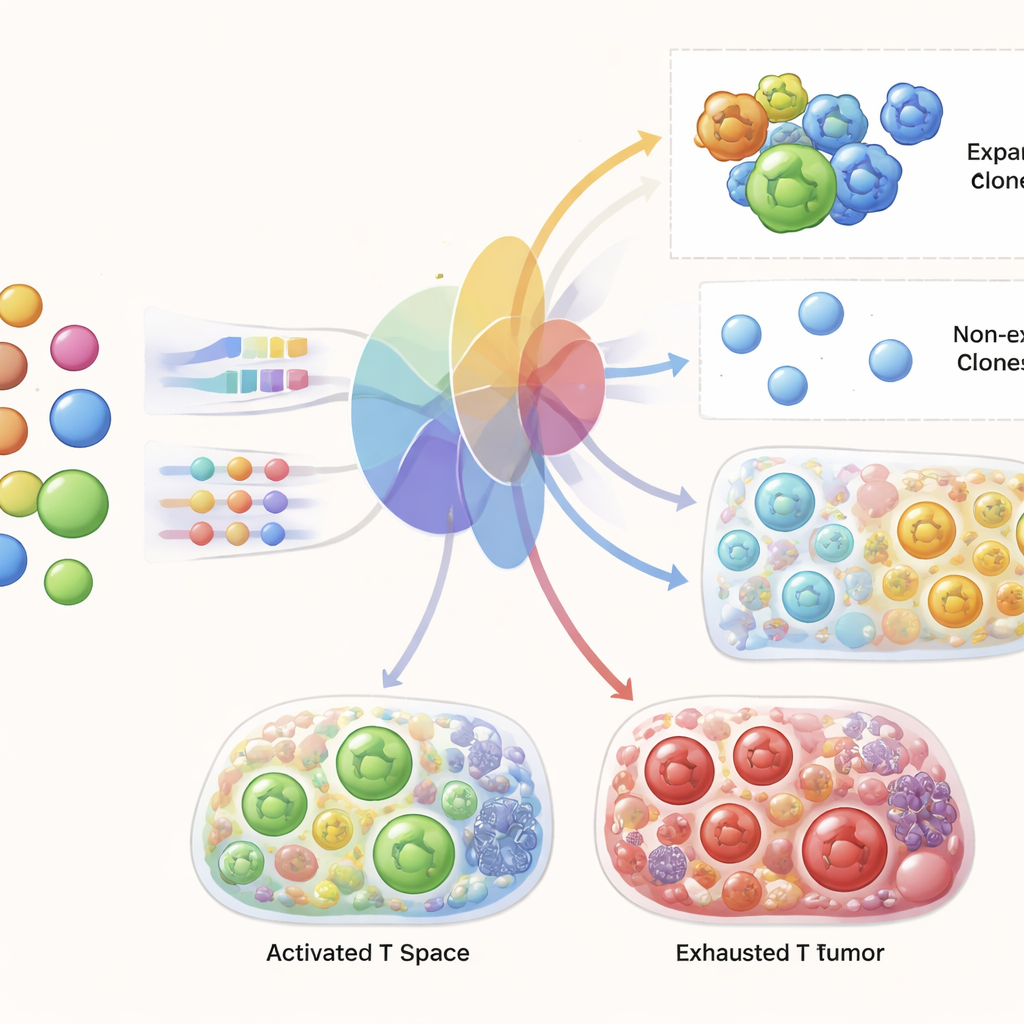
Revelando os genes por trás da expansão bem-sucedida
Como o TRIM precisa decidir internamente quais células provavelmente vão se expandir, os autores puderam sondar o modelo para ver em quais genes ele se apoia. Os genes sinalizados pelo TRIM alinham-se com características centrais da ativação de células T: reguladores do ciclo celular que controlam a proliferação, moléculas envolvidas na eliminação de células alvo, proteínas que governam ativação e comunicação, e fatores metabólicos e relacionados à migração que ajudam as células T a infiltrar-se e funcionar dentro dos tumores. Importante, o modelo identificou esses programas usando apenas dados de sangue pré-tratamento no momento da previsão, sugerindo que diferenças moleculares iniciais em células T circulantes prenunciam como elas responderão quando a terapia começar e elas encontrarem sinais tumorais.
O que isso significa para os pacientes
Em essência, este trabalho mostra que um modelo bem projetado pode aprender com dados multimodais de célula única para prever como as células T de um paciente se comportarão durante a imunoterapia do câncer. O TRIM usa amostras de sangue pré-tratamento para inferir quais clones irão se expandir, quais estados eles adotarão e quais programas gênicos impulsionarão esse comportamento, mesmo em tecidos de difícil amostragem direta. Embora sejam necessárias validação adicional e conjuntos de dados maiores antes do uso clínico, a abordagem aponta para um futuro em que oncologistas possam usar um simples exame de sangue, combinado com modelos como o TRIM, para prever a resposta ao tratamento, monitorar a progressão da doença e descobrir novos biomarcadores que orientem decisões de imunoterapia personalizadas.
Citação: He, C., Amodio, M., Ashenberg, O. et al. Multimodal framework for the joint analysis of single-cell RNA and T cell receptor sequencing data predicts T cell response to cancer immunotherapy. Nat Commun 17, 3840 (2026). https://doi.org/10.1038/s41467-026-70505-0
Palavras-chave: imunoterapia do câncer, células T, sequenciamento de célula única, modelo de aprendizado de máquina, biomarcadores tumorais